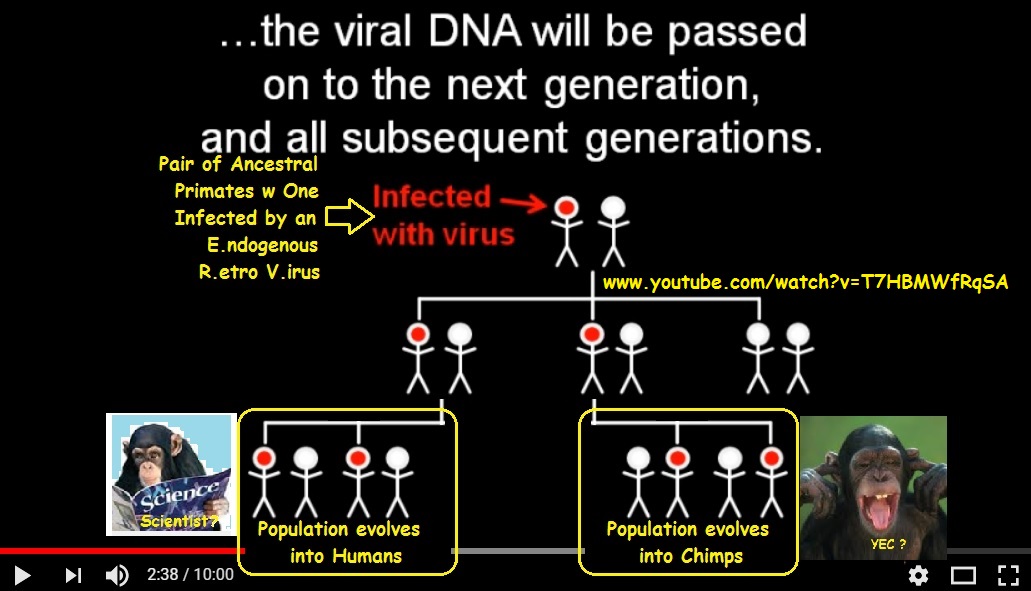
There are two good references for probabilities. First, you have a paper that looked at three different retroviruses: HIV, MLV, and ASLV. Of the three, only HIV showed any real bias, but it certainly isn’t enough to explain 99.9% of insertions occurring at the same base.
"For HIV the frequency of integration in transcription units ranged from 75% to 80%, while the frequency for MLV was 61% and for ASLV was 57%. For comparison, about 45% of the human genome is composed of transcription units (using the Acembly gene definition). "
Retroviral DNA Integration: ASLV, HIV, and MLV Show Distinct Target Site Preferences
80% of the time HIV will insert into areas that have genes in them, but about half of the genome is comprised of these areas. There are 3 billion bases in the haploid human genome, so that is an 80% chance of inserting somewhere in a set of 1.5 billion bases and a 20% chance of inserting into the other 1.5 billion. Still random enough to be used as a test of common ancestry.
The other reference is often used by ID/creationists.
" But although this concept of retrovirus selectivity is currently prevailing [37], practically all genomic regions were reported to be used as primary integration targets, however, with different preferences. There were identified `hot spots’ containing integration sites used up to 280 times more frequently than predicted mathematically [41, 43]."
Perpetually mobile footprints of ancient infections in human genome
That paper was referencing a separate paper that actually contained the results we are interested in.
“In all experiments, estimates of the m.o.i. ranged from two to three integrated proviruses per cell. A pool of 10 ug of infected TEF DNA (from -5x l0^6 cells) would therefore contain 1-1.5 x 10^7 integration events. If integration occurred at random throughout the genome (size 2x 10^9 bp) we would expect to see about two to three integrations, in each orientation, within the 500-bp stretch of DNA analyzed in each reaction.”
Distribution of targets for avian retrovirus DNA integration in vivo.
What they did was add a small bit of DNA (their DNA target) to a big pool of human DNA and then added retrovirus. They designed the DNA target so that they could detect when an insertion occurred within it, and then did the math to figure out the preference for that DNA target. Think of it like a single detector in a big field where planes randomly drop bombs. By calculating the number of bombs dropped and the area of the field you can predict how many bombs will hit your detector if it is a completely random process.
Let’s unpack the math. For purely random insertions they would have expected 2-3 retroviral insertions in their target DNA out of 10 to 15 million total integrations in the entire pool of DNA. For some DNA targets they saw almost 900 insertions, about 300 times what they would expect from random insertions. This means that out of 10 to 15 million insertions there were 900 insertions in the 500 base pair DNA target they were testing. Obviously, that type of bias is not enough to produce matches 99.9% of the time for 200,000+ independent insertions in two different genomes.
Added in edit:
Completing the math . . . even in the case of the strongest bias, that is 900 insertions out of 10 million in the same spot. This means that if every single human and chimp insertion occurred at a hotspot, only 0.009% should occur at the same position if all the ERVs in each genome were acquired independently and not through common descent. Therefore, ID/creationism would predict that 200,000*0.00009 = 18 insertions should be shared, and that is in the best of cases. That doesn’t match observations.
")