I replied to this in a previous thread. However, I think that I have a better formulation of what Hunter is claiming that I think he will feel is more accurate. In order to try to bring as much light into the conversation as possible, I am deleting my posts in the previous thread, and invite interested readers here instead.
For some reason our friend @Cornelius_Hunter loves to bring up the Klassen 1991 study, even though all but 3 of the 49 datasets demonstrates nested hierarchy at p-value << .025. Here is the diagram I believe that Hunter is referring to:
The consistency index (CI) measures the ratio of feature observations in a taxonomy that would be considered anomalous to a nested hierarchy. The blue region in the graph represents the region where we would expect 95% of the CI calculations to land, given a random assignment of features to taxa. You could refer to the blue region as the null hypothesis regime–i.e., the regime where we can draw no inferences about structural relationships within the taxonomy.
The region above the blue curve is the nested hierarchy regime–i.e., the area where scientists would feel comfortable inferring the existence of a nested hierarchy structure in the taxonomy. Of course, it is useful to ask how confident the inference of a nested hierarchy signal is. Here, the diagram is a great help: the confidence is proportional to the distance from the blue region. The greater the distance, the more confident the inference of nested hierarchy can be.
But we can do better. The statistical significance of any point in Klassen’s scatter plot can be approximated by comparing the vertical distance from the blue region to the vertical size of the blue region at the intersection point. Half of the blue region’s vertical size represents two standard deviations from the mean expected by random assignment of features to taxa. Since the standard deviation is scale-invariant along the Y-axis, we can use the diagram to approximate the number of standard deviations between an observation and the randomness (null hypothesis) mean.
Here is an illustration:
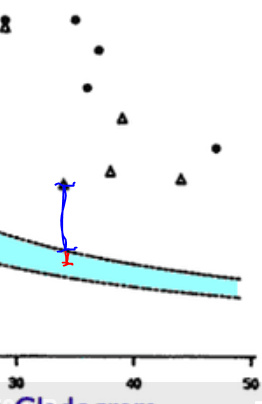
The red line segment represents about 2.0 standard deviations from the randomness/null hypothesis mean. The blue line segment is the distance of a particular observation–the one which is closest to the null hypothesis regime–from the null hypothesis regime. The ratio of the blue segment to the red segment appears to be about 4::1. This means that the observation’s total distance from randomness is about 10.0 standard deviations, i.e., 2.0 SDs along the red line segment and 8.0 SDs along the blue line segment.
Under the null hypothesis, the probability that an observation could lie at a distance of 10.0 SDs is 0.00000000005. Stated another way, the probability that this one taxonomic observation reflects a nested hierarchy rather than randomness is 0.99999999995
Adding even stronger evidence for nested hierarchy in biological taxonomy is the fact that this one observation is one of the weakest in the 49 datasets studied by Klassen. Other observations lie at distances of 20 or even 30 SDs from the randomness mean.
Finally, it is appropriate to take all of the observations as an ensemble. When the vast majority of the observations show the existence of a nested hierarchy with extremely high probability, and only 3 observations with tiny numbers of taxa are in the randomness regime, then the conclusion of nested hierarchy is inescapable.
Unless…
You take Hunter’s approach, which asserts that the existence of any significant amount of noise is evidence against evolution. Hunter stated the following about Klassen 1991 in an old different thread with @Swamidass:
Hunter is not arguing over the statistical significance of the hypotheses, he avers. As far as I can tell, he is instead arguing for a Bayesian analysis in which the evolution model products a Consistency Index of 0.8 for all taxonomies, a Design Model predicts a Consistency Index of 0.6 for all taxonomies, and a randomness model predicts a Consistency Index of 0.15 for 30 taxa. (These are the only numbers he has ever supplied in the Signal vs. Noise Part 1 thread, as far as I can tell.) Since the CI scores are closest to the design model, he asserts the design model should win the model selection contest.
Hunter’s Bayesian analysis suffers from four flaws, all of which are fatal to his argument:
(1) The theory of evolution is a stochastic model, so it predicts significant noise.
I have addressed in another thread how the noisiness of stochastic models need not erase a signal, and does not do so in the case of the theory of evolution. I won’t repeat myself.
(2) The mathematical models of evolution predict more noise when more taxa are included in a taxonomic analysis. You can see this by inspecting the blue curve in Klassen’s diagram: increasing the number of taxa decreases the expected Consistency Index for a nested hierarchy.
(3) Per our friend Joshua @Swamidass, computational biologists have quantified the expected CI for taxonomies. The expected value is not the uniform 0.8, but it does conform quite nicely to the observations.
(4) Hunter has been unable to provide any quantitative estimate of the amount of noise predicted by the design model. In the absence of any quantification of predicted noise, it is impossible to predict the CI of the 49 taxonomies under the design model. Since the design model can make no quantified CI predictions, it cannot be included in the Bayesian analysis.
I assert this because I asked Hunter point blank in another thread:
And here was his reply:
Evidently Hunter does not know.
Hunter cannot make any mathematically-derived statements regarding noise expected under a design model, so it is impossible for the design model to predict how much inconsistency to expect in taxonomies. If the design model cannot quantify how much inconsistency to expect, then the design model cannot make Consistency Index predictions, full stop.
Conclusion
Perhaps, some day, the design model will be sufficiently elaborated in a mathematical way to include it in a Bayesian analysis of Confidence Interval data. If and when that day arrives, who knows the result will be? But that day has not yet arrived. Until it does, it seems premature for Hunter to make any claims based on Klassen (1991) about the supposed inferiority of an evolutionary model to a design model.
Best,
Chris
