At a genetic level we see a great deal of similarity between organisms. This similarity is a fundamental feature of life, that has to be explained by any theory.
Evolution explains that this similarity as largely caused by shared history, by way of common descent. We see function often aligning in sensible ways on phylogenetic trees of sequences and organisms. Of course, this is not perfect, because we also know that life changes over time. As the YEC Walter ReMine accurately points out in The Biotic Message, function will not always follow a nested clade pattern in an evolutionary model. (He argued that YEC would always follow the nested clade pattern, but it turns out that biology does not always do so).
Anti-evolutionists in the ID and creationist camps explain this similarity as the caused by common function engineered by a common designer. The thought here is that any shared function we see aligning to phylogenetic trees is just an illusion. Trees are constructed by similarity measurements, and these measurements are shaped by shared function. Nothing is gained by constructing a false history through the phylogeny, and we would do better to just see similarity as caused by shared function, shared design.
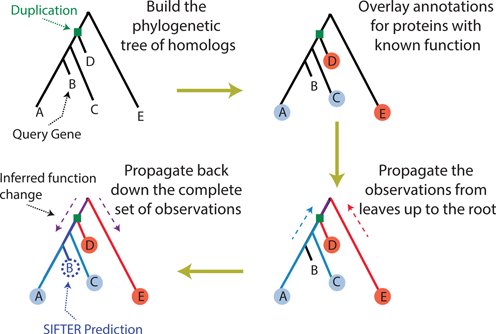
So there is a field of bioinformatics that directly tests these two hypothesis for the immensely practical problem of determining the function of unknown sequences. The experiment is straight forward. We compare the accuracy of function prediction algorithms that use pairwise similarity (e.g. BLAST searches) to assign function with those that use phylogeny instead (e.g. SIFTER).
We find that phylogeny informed predictions are two times better than similarity informed predictions. This is a fairly direct test of the two hypothesis. If phylogenies are an illusion, that do not actually represent a true history, why do they predict function better than similarity? I would say this is evidence that phylogenies are more than just an illusion. They capture something about function that is more informative than just similarity, even though they are inferred from similarity.
So similarity is used to infer phylogenetic history, and this history is more correlated with function than similarity itself.

You can read some of the references (and the figures are great too) here:
And follow me (@swamidass) and one of the early pioneers in the field (@phylogenomics) on twitter for an entertaining dialogue on this.
