Ha! Exactly what I did at first, at the gene level, and showed DAGs produce distinct trees just like genealogical processes do. The feedback was I needed to do this at the nucleotide level with official tools.
Now, I’m not very familiar with the PAUP software, but I think this addresses what you want. I create 100 random trees, run a likelihood test and sort the trees. If the clade structure is the same, then the sorted range should be pretty small, and visa versa.
There is a large DAG dataset that has 26 taxa.
There is a smaller DAG dataset with 18 taxa.
For comparison I have a primate dataset, with 12 taxa.
For the primate dataset I have a high score of 7300 and low score of 6394. For the 26 taxa DAG dataset I have a high score of 6768 and low score of 5857. For the 18 taxa DAG dataset I have a high score of 7816 and low score of 6915. The spread is the same in each case, but the larger DAG dataset has a lower overall score, which is to be expected with more taxa. So, there is not a clear difference between the clade structure in real world data and in the synthetic data, at least with regard to CI. I echo @Chris_Falter’s question, is there a different metric you recommend in place of CI? Hopefully a simple one
@Chris_Falter, for reference, here is the same post over at theskepticalzone.com, where you can see the evolutionary biologist weigh in with their opinions.
Some of the comments are both helpful and incisive, such as corneel’s and Allan Miller’s. Others were more emotive, but even so they point to ways you could improve your presentation. For example, your discussion section could perhaps benefit from a less combative phrasing while still making the same points.
BTW, how do you know who the commenters really are? Is “Flint” a tenured evolutionary biologist, or some random guy/gal? And how would you have confidence in your assessment?
Because I do not really know who the commenters are, I would want to see your paper proceed from informal review at TSZ to a legitimately peer-reviewed publication. (There are plenty of vanity pubs that aren’t really peer-reviewed, not that I know how which ones are good and which are flotsam and jetsam in the realm of biology.)
Or just maybe, thousands and thousands of biologists, geneticists, etc. who’ve spent not just a year out of curiosity but decades upon decades of testing ideas and building technologies on the ideas we’ve figured out so far about genetics.
First, it’s a bit of a strawman to suggest that high CI = high support of nested hierarchy. It’s nowhere near as simple as that. For a start, the CI was developed for (binary) morphological data, and it’s not employed on nucleotide phylogenies precisely because it’s not very informative about the quality of the tree.
The CI is sensitive to many different factors in the dataset, and one that appears to be very relevant in the DAG case is the number of gaps in the alignment necessitated by the massive variation in leaf sequence lengths. This produces a highly uneven distribution of character states, inflating the CI. https://onlinelibrary.wiley.com/doi/epdf/10.1111/j.1558-5646.1989.tb02626.x
(note especially pages 4-5/15)
In both cases, the alignments are obviously completely unlike any seen in rigorous phylogenetic analyses of homologous DNA sequences - i.e. unlike what we see in reality. Extremely low branch support values are consistently found in phylogenetic analyses of random or DAG-based sequences, unlike in analyses of homologous sequences. Mimicking one summary statistic (that is already known to be sensitive) is far from demonstrating that DAGs (or anything else) account for observed data better than an evolutionary process following a nested hierarchy.
Before I forget, who I am: I’m currently teaching at a small university; research focus is on mollusk classification and evolution, so I did calculate consistency indices back in the 1990’s; theological background is Reformed. These days (i.e., in the past couple decades), the consistency index is not used much at all. It is affected by the number of taxa and the number of organisms. By definition, a star tree will have no inconsistencies because it does not indicate any groups that could be inconsistent with anything. So a consistency index is not helpful in assessing a star tree versus a more resolved pattern. In an analysis of a large data set, there are likely to be a very large number of trees that do not differ much in CI. Klassen et al. 1991 (Consistency Indices and Random Data on JSTOR) argued that finding that the CI was above that expected for a random data set of similar size was a useful indicator that there is phylogenetic signal in the data set. But people don’t generally perform phylogenetic analyses on data sets unless they think there is phylogenetic signal, so that’s not very informative. The g1 statistic suffered a similar fate - it measured skew in the tree distribution, but any real data set of interest to phylogeneticists had a reasonable g1 value, and so both of those metrics gradually fell out of use. Instead, analyses focus on the use of analyses such as maximum parsimony, maximum likelihood, or Bayesian analyses to determine what are the best trees. Analyses such as bootstrapping, jackknifing, and decay indices give a rough picture of the degree of support for a particular clade. If the support is strong, that would indicate that the data strongly favor a particular clade versus the alternatives; non-evolutionary ways of generating data (or high enough evolutionary change to have effectively randomized the data) would not be expected to generate much higher support for some groups than others. For example, under a non-evolutionary scenario, taxa A, B, C, and D could each have roughly equal numbers of similarities for each of the others. The ways that A and B match each other would not be expected to necessarily also show up in other pairings - if A and B are similar, in a non-evolutionary scenario, that would tell us nothing about whether similarities between A and C should also be found in B. Another key test of evolution would be whether the patterns found from one data set for a given set of taxa are also found when a different set of data (such as other genes or morphology versus DNA, etc.) is analyzed. Some random variation and homoplasy is expected, but not a complete lack of correlation.
I am less interested in the pseudonym commentators, and more interested in Harshman’s comments, and potentially Felsenstein if he deigns to comment. There are a couple known academics on that forum, the same ones that frequent PS, and perhaps one or two more. That being said, the pseudonym commentators can at least point out specific deficiencies in my analysis, which will be helpful. If my work makes it through a couple forums, then I’ll consider peer review.
I am not so sure making something mimic what we see in reality is the relevant criterion here. The question is whether these metrics are good indicators of genealogical processes, or whether the metrics are also easily swayed by other generation processes. If the latter is the case, it isn’t clear they are good indicators of genealogical processes.
However, if making it ‘look’ like normal alignments we see in the wild is a big deal, I can do that too. It will not be a big adjustment to the simulation. We will see if it makes any difference.
What I would really, really like to find is something quantitative, whether it be a metric, an algorithm, just something, that is supposed to be a reliable, quantifiable indicator of whether evolution has occurred. If you know of a specific such thing, please let me know!
That is exactly what I did above. I showed DAGs produce CI way above the chance cuttoff.
Did you see my response where I did this? Here it is again:
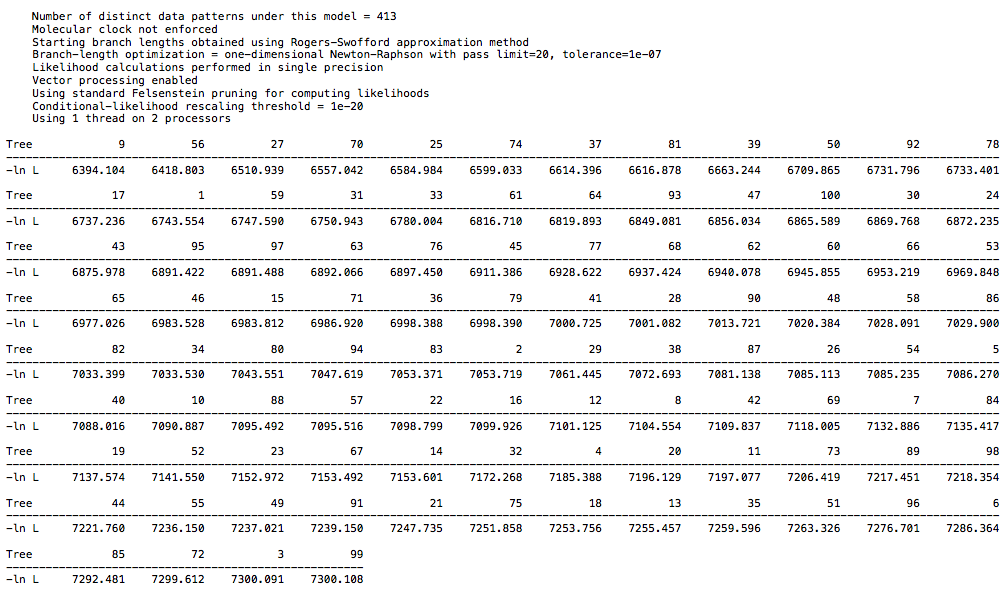
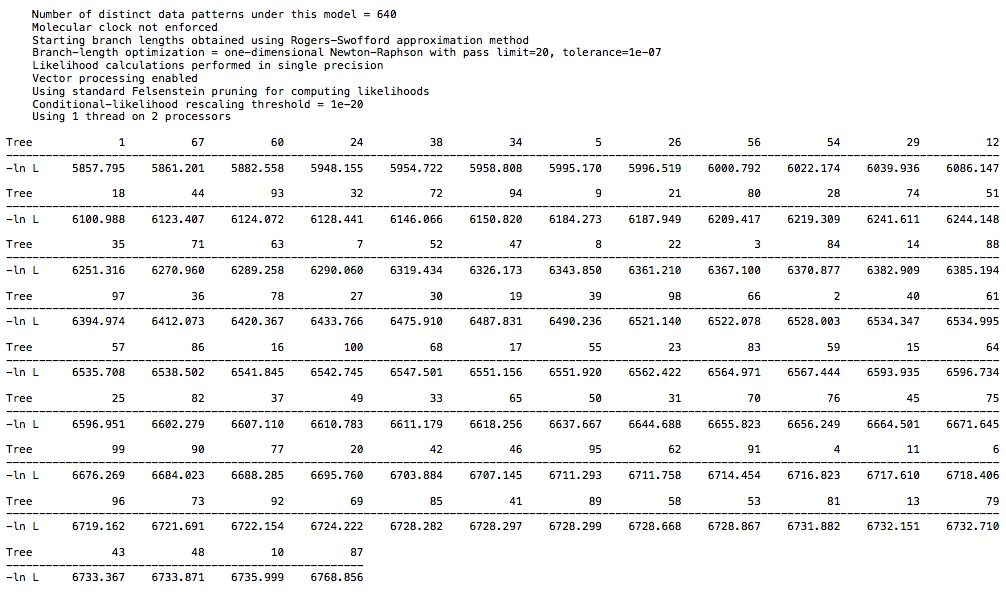
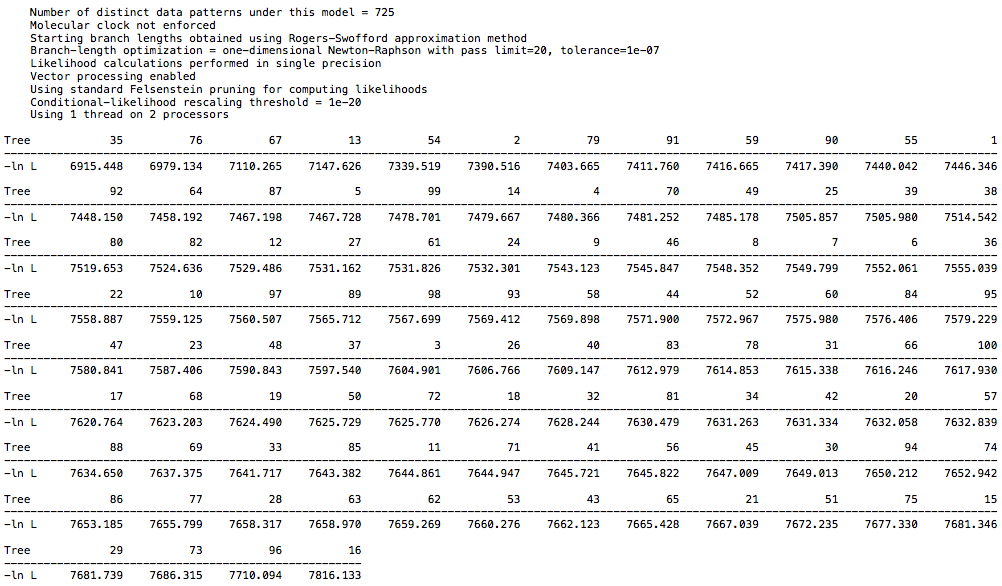
I ran a likelihood analysis on my DAG dataset and compared to a real world dataset, and it was indistinguishable.
Anyways, can you recommend a specific process/metric I can use that is supposed to reliably indicate an evolutionary process created the dataset?
I had thought this CI thing was a really well established result from the Klassen 1991 paper that analyzed 70+ CI results. But now you are telling me that CI metric is meaningless. So, what do we make of the Klassen 1991 paper and their analysis of 70+ CI results? Is that also meaningless?
The field of evolutionary science is really confusing!!!
That would be a start, but if you want your results to be meaningful and be able to interpret them accurately, you should try reading some of the numerous papers discussing merits and pitfalls of the CI, rather than taking it to be either an infallible “magic number” or utterly meaningless. You should also recalibrate the significance cutoff if you want to use nucleotide data, bearing in mind that the random data generated by Klassen was binary morphological data. It does make a difference.
I imagine many things would be confusing if you make no effort whatsoever to understand them. It’s almost as though reading one paper isn’t going to give you a detailed understanding of a subject in a field you’re mostly unfamiliar with.
I’m just going off what the Talk Origins page literally says. Perhaps I am too literal in my interpretation, and there is a lot of unstated qualification.
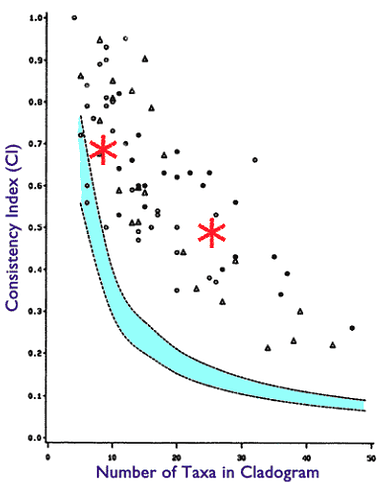
phylogenies based upon true genealogical processes give high values of hierarchical structure, whereas subjective phylogenies that have only apparent hierarchical structure (like a phylogeny of cars, for example) give low values
One widely used measure of cladistic hierarchical structure is the consistency index (CI). The statistical properties of the CI measure were investigated in a frequently cited paper by Klassen et al . (Klassen et al . 1991; see Figure 1.2.1). The exact CI value is dependent upon the number of taxa in the phylogenetic tree under consideration. In this paper, the authors calculated what values of CI were statistically significant for various numbers of taxa. Higher values of CI indicate a greater degree of hierarchical structure.
I create a simulation where a non genealogical process creates data that measures a very statistically significant level of CI, thus directly falsifying the literal reading of the above.
Again, I cannot address any unstated qualifications to the above, all I can address is what is stated literally, and as such it is provably false.
As for the binary morphological data, that was my first approach, and it scored even higher CI values that were way outside the published values for the various number of taxa. This nucleotide approach was merely to address the criticism that I had not done my analysis at the nucleotide level. Now that I’ve done both I rest my case.
You are being too literal. Given that the article specifically cites the Klassen paper, which shows the type of simulated data that generates low CI in contrast to real data, it’s not hard to understand the intent.
As I said earlier, one major reason you are getting high CI values is because the DAG nucleotide datasets, by their very nature, contain vast amounts of missing character information in them, producing extremely uneven character distributions. This is totally different to realistic sequence data.
If you want to argue that DAGs are a better explanation of what we see in nature, you need to get DAGs to match all the relevant characteristics of that data, not just a single one while disregarding all the rest. Produce a DAG-derived dataset that produces high CI and simultaneously mimics real-life data in its properties, then you might have an interesting result. Simply exploiting known limitations of the CI using a Lovecraftian monstrosity of a dataset isn’t impressive.
Any tips on what these properties are? The only one I can think of that I’m missing is the genes have big gaps between them during the alignment. That’s simple to fix, and I’m doubtful it’ll change the result. Otherwise, I’ve addressed the criticisms I’ve received here and in other forums:
use a standard metric - I used CI
use a standard null hypothesis - I used Klassen 1991 CI cutoff values, and plotted my results on their chart
show the tree is well supported - I ran PAUP’s bootstrap and produced well supported branches
show the random trees have significant divergence in structure comparable to real world data - I ran such a test comparing to a hominid dataset and posted my results higher up in this thread, showing my results have the same range of structure as a real world dataset that we are most confident has been produced by a evolutionary tree
create a nucleotide level analysis - done, per our discussion here
Let me know if you can think of any other properties I’m missing, or if you can recommend some papers. I’ll keep examining the literature.
Even better, show me where in the literature someone has undertaken a similar analysis to my own, running standard tests for phylogeny on DAG generated datasets. I’ve not been able to find any such study, so it appears my work is the first of its kind.
That’s the biggest one to solve up front. After you’ve done that, we can look and see what the other problems might be.
Do you understand why you can’t compare results from Klassen’s binary datasets with these nucleotide datasets? Playing around with different datasets it appears that simply going from a binary dataset to a quaternary dataset, keeping everything else the same, produces ~2x higher CIs.
Can you post the original data you used there?
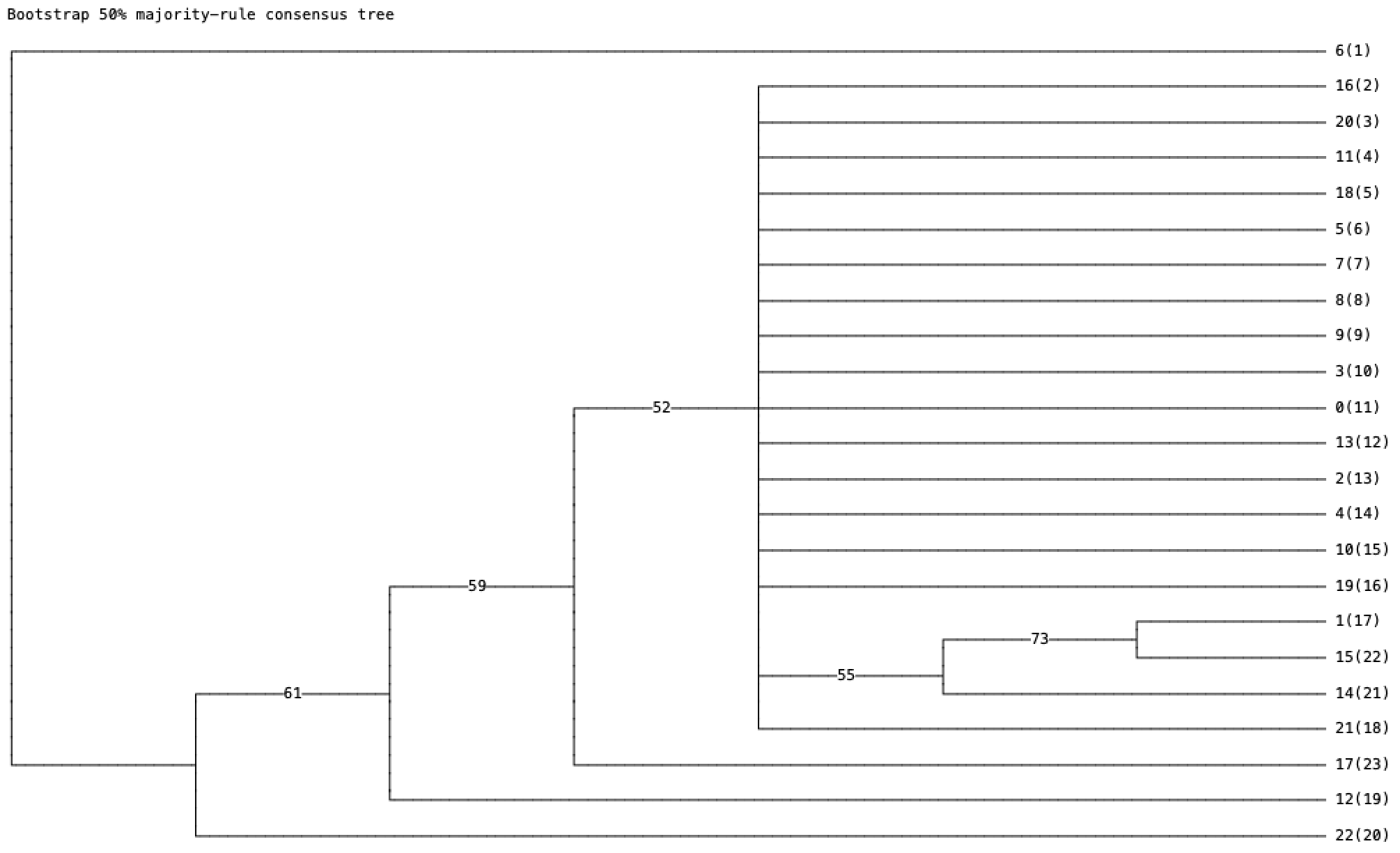
One other thing it’s worth pointing out is that you’re not actually performing a phylogenetic analysis in PAUP, based on how you’ve described your steps. You load the nexus file then click “generate trees”, but this simply generates 100 (by default) random trees based on the data. When you then click “describe trees” and select tree 1, this is just describing the first of 100 random trees. To generate a rigorour phylogenetic tree, you need to laod the nexus file, then use one of the search options in the “analysis” tab. You’ve said that when you use the bootstrap option it asks you if you want to increase the number of trees, and that you click “no” because otherwise it runs forever. This shouldn’t happen. It’s simply asking you if you want to increase the number of trees saved, and there’s no reason to do that in this case, so clicking “no” is the correct choice, but the analysis doesn’t run forever, it just runs until the number of bootstraps you chose have been completed (default 100). On my computer that takes less than a minute with these small datasets.
I certainly don’t get well-supported branches when I run bootstraps of DAG-derived trees. Here’s one I got as an example:
Maybe I don’t understand what you mean by a binary morphological character. I think you mean something like ‘has wings’, which for some creatures is true and others is false. We have an array of such properties, and each creature gets a string of trues and falses, or 1s and 0s, representing whether they have each particular property. If this is what you mean, then as I said, this is the first analysis I did, and the CI values were even higher than what I get with this DNA version.
Can you post your settings so I can reproduce what you’ve done? For some reason I can consistently get well supported trees with my datasets. My settings are probably wrong.
Also, the question is not ‘are there DAG datasets that produce bad trees?’ Rather, the question is ‘are there DAG datasets that produce well supported trees?’
Some of the DAG datasets are indeed pretty poor, but others seem to produce well supported trees.
This may indeed be the fatal flaw with my approach. I don’t know what counts as a ‘high’ RCI value. Do you know the range of ‘high’ RCI? The values I see from my datasets are pretty small, less than 0.1.
The fact that you did this analysis is unrelated to my point here - you can’t plot results from quaternary data on the graph from Klassen that used binary data. I suspect your binary results suffer from the same problem as these - a huge amount of “missing” data.
What I did:
Drag the DAG nexus file into PAUP
File>execute ‘filename’
Analysis>bootstrap/jacknife
Leave all the defaults (Bootstrap, 100 replicates, random number seed: 0, Full heuristic, Retain groups with frequency >50%)
Click Continue
Leave all the defaults (get starting trees by stepwise addition, swap on best only when multiple starting trees exist, stepwise addition sequence: simple, hold 1 tree at each step, TBR swapping algorithm, reconnection limit = 8)
Click Search
When the Maxtrees query comes up, click no
Wait a few moments for the analysis to run (see progress in top right of window)…
Then the tree is output
That’s it.
Right, so I should point out that you shouldn’t be using likelihood to assess how well a tree is supported, as you did earlier in this thread comparing DAGs to the sample primate mtDNA dataset. Once again, the very nature of the DAG datasets you’ve produced is that they contain huge amounts of gapped sequences. These gaps get labelled as “missing data”. PAUP calculates likelihood based on the total likelihood of each site. “If this phylogenetic tree is correct, what’s the likelihood that the states of these taxa in site X would be the same as what is actually in the dataset?” This question is asked and answered for each site. If most sites are missing data from most taxa (as they are in your DAG alignments), then the dataset becomes readily compatible with a huge number of trees, rather than a small number. This is reflected both in the poorly-supported nature of the final phylogenetic tree (resulting in massive polytomies), and in the fact that the “best” tree has a likelihood extremely close to a random tree. If the tree is highly ambiguous, then almost any dataset can get a high likelihood of fitting to that tree.
Load a DAG nexus file, and generate 100 random trees. See what the likelihoods look like (in my run, they were all around roughly -4400-4450 (ln L). Then perform a bootstrap analysis to arrive at a single “best” tree, and see what the likelihood looks like for this specific tree. I got a likelihood of -4435 (ln L). It’s well within the range of random trees for this dataset.
Contrast this with the primate dataset. If I generate 100 random trees from it (as you did above for some reason), I get likelihoods of around roughly -6800-7200. After performing a bootstrap to get a single best tree (which takes orders of magnitude less time to process than the DAG dataset (0.05 seconds as opposed to 65 seconds BTW, that’s a clue), the likelihood of this tree is -5992. A significant increase in likelihood compared to random trees. You can’t compare likelihood scores between different datasets, so it’s irrelevant that the DAG tree likelihood is lower than the primate tree likelihood.
This is all because the primate data points towards a single well-supported conclusion, while the DAG dataset doesn’t because it wasn’t generated by a tree-like process.
I don’t know in detail, and it is probably still going to be affected by some of the same biases that affect CI, but it seems to buffer it. For example, in a sample “best” DAG tree I got a CI of 0.84 and RCI of 0.18, compared to a CI of 0.65 and RCI of 0.39 in the “best” primate mtDNA tree from the dataset you used.
A higher CI for a tree of the same type based on the same data indicates that the tree better reflects the patterns in the data. Thus, the fact that trees resulting from evolutionary analyses characteristically have higher CI than random does suggest that an evolutionary model is a decent approximation of how the underlying data were generated. But you can’t say “here’s a CI of 0.93, so it’s good”. It would have to be compared with random results for the same data set and tree-building approach. And even then, “better than random” cannot be equated with “good” - there may be better trees not analyzed. Likewise, the CI for a tree with many polytomies is not comparable to that of a tree forced to be fully binary.
TalkOrigins isn’t the greatest source; it does have plenty of good information but not all the arguments are sound.
I’ve updated the simulation so it achieves these metrics. The thing I cannot reliably reproduce is how well supported the branches are in the primate dataset.
Here’s the updated simulation. You’ll most likely need to run it a couple times to find a high scoring dna.nex file. I’ve simplified the graph visualization so it is much easier to understand.
You can use this online graphviz to view the graph. Just paste in the contents of graph.dot.
The support for the branches in the mtDNA data set indicates that a branching tree evolutionary model works well for the data, giving definite support for some configurations and not others. Random data (whether it’s simulated data or generated by an evolutionary process but too highly changed to retain clear traces of its origins) will not give good branch supports because different tree configurations will all be about equally as good a match for the random data.