I made them up and have no bearing on the actual generation model for the dataset. Each of the examples I gave above shows how the dataset is actually generated (DAG and random sampling), and then the subjective phylogenetic tree that is derived from that dataset. To create the phylogenetic tree I use the UPGMA algorithm on the dataset.
I am happy to explain anything I state in further detail.
Alright, I can tweak my code to be alignment based, if you think that’s the missing ingredient to make the CI dependably signal phylogeny. We shall see.
I don’t have time to figure out that PAUP software, unfortunately.
Pretty much. It is well known that the scientific method does not constitute a rigorous logical proof.
It is based on what is reasonable. When a procedure gives the same results a thousand times then it is reasonable to believe that under the same conditions it will give the same results the next time also.
And no matter how much you point out that this is not logically rigorous it will not change the fact that expecting a different result is unreasonable.
Yes that is the basis of scientific honesty which is vast improvement over the rhetoric of lawyers and salesmen who seek to prove their proposition any way they can make sound good.
And the result of many such tests is a bit more than just affirming the consequent based on one conditional A->B. It is more that every test from every conditional you can think of gives the same answer.
A->B
A->C
A->D
A->E
…
All these consequent are tested and verified to be the case, and there is not even one case where a consequent was found to be false. Then in science we think it is reasonable to conclude that A is the case, though we may say something like… to the best of our knowledge A seems to be the case. Though we keep on thinking up new tests and trying those too.
Here’s a simple counter example to alignment. Let’s say we have the four taxa:
GA
AT
TC
CG
Different letter in each position, so number of characters is 8.
You can see the taxa are created by cycling through the GATC letters, not evolution. But, we can run an alignment algorithm to create a tree of 6 steps:
null
|-A
__|- GA
__|- AT
|- C
__|- TC
__|- CG
In this case, the CI is 8/6 = 1 1/3, even greater than 1. We can retroactively say there are only 6 characters, due to the alignment derived, and bring the CI down to 1. In either case, we have a non evolutionary process that generated taxa with perfect, or even better than perfect, CI score using an alignment based approach.
I would strongly suspect that you need a minimum amount of homology for these methods to work. Again, I’m not an expert but I wouldn’t expect 4 completely different sequences to be a proper data set.
1 Like
Klax
(The only thing that matters is faith expressed in love.)
27
Evolution is a rational and scientific fact that needs no evidence whatsoever from genetics. Darwin had none.
Well, I am still working on getting PAUP software to work. On the face of things, it seems trivial to produce high CI trees from purely random sequences, since I can align the random sequences with edit distance, gap anything that doesn’t align, and then build trees from what remains. But, still gotta unravel the NEX file format to be able to test this out.
And success! I created 20 random 80 character sequences sampling uniformly over GATC. Here is the Python script.
from random import randint
strlen = 80
num_seq = 20
for i in range(num_seq):
print(">", i)
print("".join(["GATC"[randint(0,3)] for _ in range(strlen)]))
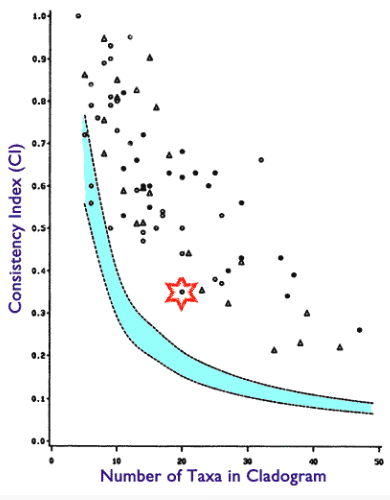
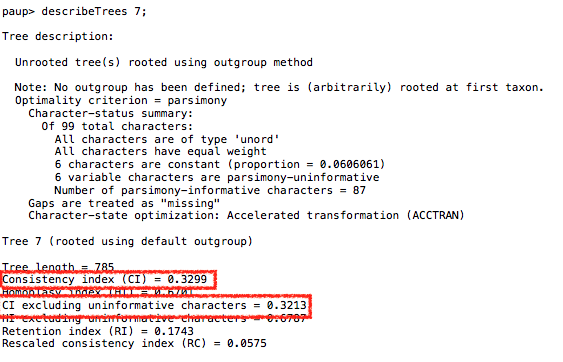
I then performed multiple sequence alignment on them with ClustalW, which produced the following alignments. When I use the PAUP software to create trees, the trees consistently get a CI score of 0.32-0.33, which is well above the threshold in the above chart, and ventures into published CI values.
So, since I can generate phylogenetic signal CI from random datasets, this means the phylogenetic signal cannot tell us if a dataset exhibits common descent.
The phylogenetic signal argument for evolutionary common descent should be retracted until it is revisited with much more rigorous controls.
I used the PAUP software downloadable here: PAUP* Test Version Downloads
I used the online ClustalW here: Multiple Sequence Alignment - CLUSTALW
You can just paste my below sequences into ClustalW and it’ll perform the alignment. Using PAUP is more complicated, since you have to mess with the NEX format. Once you do that, push the ‘Trees->Generate Trees’ menu option and push ‘OK’, and then push the ‘Trees->Describe Trees’ menu option and pick any tree and push ‘Describe’. It’ll print out something like:
For anyone wishing to reproduce my results, here is my original dataset in FASTA format.
> 0
GGCTTTAAGTCCGTCTCGGGATAGGTTAATAAACACTGCGCCGGTCCTAGCAATATGGCGATGCATCTGGTCCACGGCGC
> 1
TACGCTTACGGAACACGGGCTATCGATTTCATTTCGGTTTCTGCGGTGCATTCCAAATTCTGAAATGTCACTTCGAGTAA
> 2
GAAGGAGGTACTGTATGTATGTCCGCGCTTACCACTGGCAGTACTTGTTCCCGTTTCACGGCGCCCATACACGAGCATGA
> 3
CATTGGAACGTTATAGGTATATCCAATGACTTGAGAGACTGCCTTGTGATGTAGATCCGCAAGACCTCGGTTATTGGATT
> 4
TCTCCGAACGTCTGAGTGACGCTTATTAGTACATCCAAAGTTTGGCTAAGCGCAGCGGACATCCCAGTTACTCTAAAACT
> 5
ACCAAGGACCGTGCACGACTATTTTCGACCTCTTTTCCCCTACGATAAGGTCCACCCGCGCTACTCGAGTGACAGCAGAT
> 6
TTGCCCCAGCCACTTAAACAGTATTCCATTTGGACTTCCTACCAGGTCACACCTTTCCCCGAAGCCAACCTGCGCTGGTA
> 7
TTGTCCGACCATGCGTGGAGAATGAGTATACTAATTGCGGGGAGGCTTAGGAGTATACATACGCAACGGATGACGCCGAG
> 8
CCCAAACCCATCTAATGGTGGCTGAGACACACTTAGGAACGACATTTCTGGTAGTTACGCGAGCTGACAAGCTTGCACGC
> 9
TGGTTTTTAAAGCTACACCAAATCCTAGACCTCATTTATAGTGGAGGATAATTAGACTAGTTCGTACAGGTCCGATTACT
> 10
TGTCGCTGCAGGCACGACTTGGAACCACCAGTGCGTGGTCACCCGGGCTGTTCTTGCAACCTTCCATGCAAGATCTCTAA
> 11
CATCTTCGTCCTTTCATCTCTTACTAGTACGTGTCTAGGCAGGGTCTGCCTCACTGATAGCCGTCCGTTTTTTCGACAGT
> 12
GAGCTTCTGTCGCGTCTCAAGAATAAAGCGCCAATTAACGTACAGCTGGCCCCTGACGGTAATGAACGACGAAAATGCGG
> 13
GGGCGGTGCCAATTGTTCCAATCATTGTAGGGGCTCGAGATATCGCTCGTCTCATTTCGCTATTCTTTTATTTAGGGACG
> 14
CAAGTTGGTCTAAGTGTCCGTTGATTACGGCGCCTTGTGCCCATGCTCGCAAAATGCGGCGCGATGCTCGACGCGTAGGG
> 15
AAGTTCGCCTATAGGGTATCCAATGCTCTCGTGATCACCACGACCTAATCGCACTCCTGTTCGTTACTACGCCCCTTCTG
> 16
CCTAGTGAGTCGGCCGCGAGCATCCATATCAACAATGCAGATATAAACCTCCCGAGCGCTCCTAAGATACAGATTTAATC
> 17
CCACGGTCACACATGCTCTGGGATCCACTACCTCTTAAGCCCACAACTAAGCAAAGCGCAGGGTATTAATTAGCCTCATA
> 18
GGTAGGGCCCATGATATCCGCAGCTCGAGTGGCATTTTAGCCACGATGCATCAAAGCATCAGAAGGATCCGCCCTTCACT
> 19
GACATGTTCCCACGTGTGGATCATACAGTTATCAGACGCTAGTAAGGACGGGACAGGTAATCAAGGCTGGTCTGCCTCTA
And here is the NEX file. You can use this verbatim with PAUP to get my results.
@T_aquaticus@Chris_Falter here’s an example of sequence level analysis of the DAG using standard alignment and tree generation tools ClustalW and PAUP. I modified my previous DAG expriment script to replace the gene ID numbers with a random sequence generated from GATC of 20-30 letters. Then I process the resulting sequences with ClustalW and then PAUP to generate trees and calculate the corresponding CI.
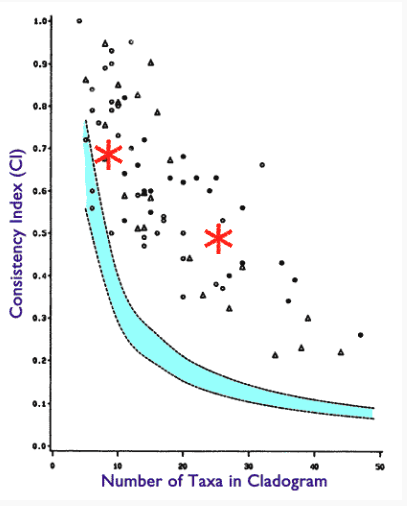
For 10 taxa I get a CI of 0.68 and for 26 taxa I get a CI of 0.48. These are well within the values achieved with studies of actual data, see following chart, which again shows high CI values do not tell us whether the dataset is the result of common descent.
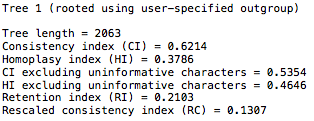
Thanks. I added an outgroup random sequence and that appears to have increased the CI score.
A number of weeks ago I stated that a DAG will generate a phylogenetic signal just as well as an evolutionary tree. You requested I conduct this analysis at the nucleotide level to demonstrate the claim, and I have done so. At this point, I believe I have made my case.
I respectfully request this forum decline to use the phylogenetic signal as an argument for evolution, at least until it is rigorously demonstrated how to eliminate DAGs as an alternate models for real world datasets.
I believe this careful approach fits well with the Biologos mission, since we want to show religion is consistent with good science, not anything cast under the heading of ‘science’ whatsoever. As such, if we happen upon a purported evidence for evolution that does not hold up under scrutiny, it is best to lay it to the side until we reach a more rigorous demonstration of the evidence’s veracity. This is important, because if we wantonly associate ourselves with any and everything called ‘science’ we may associate the Biologos brand with something that becomes publicly discredited, which will in turn cast a shade on the Biologos brand.
I respectfully request that you run your analysis through the scientific peer review process before you make overreaching demands of your fellow forum participants.
What do you know about the foundational work and testing on phylogenetic signals that was done prior to your arrival? So far you have not cited a single source.
And this: The fact that you as a self-professed “armchair” scientist think that your unreviewed work gives you the standing to overturn decades of hard work by thousands of biologists is astonishing.
I’ve cited multiple sources, primarily the Klassen 1991 chart that the forum participants rely on in all the ‘nested clade’ arguments.
The claim you and others make is phylogenetic signal is strong evidence for evolution. I’ve conclusively proven this wrong. You can provide some alternate evidence to fix your case. Otherwise, your argument is defeated. Simple as that.
You are talking to an armchair forum participant who thinks it wise not to rely on fellow armchair participants. Instead, I rely on a community of experts who have a well-proven practice of mentorship and peer review to establish what is sound and what is not.
Now you may or may not have something important to refine and possibly publish, Eric. I have no clue what the true state of the matter is. As I have already indicated, I would love for you to go talk with John Harshman at Peaceful Science, and whoever else he might refer you to. Your curiosity and your work ethic are laudable, I will grant you that. The problem is this: I have no way to tell whether your work is valid or, in the alternative, based on a fundamentally flawed simulation.
If you, as a fellow armchair participant, are not interested in working with the trustworthy members of a trustworthy community of experts, then I have no reason to ascribe any probative value to your work.
Well, I’ve laid out in great detail how it all fits together. And what I need to show is not very complicated. To disprove the nested clade argument all I have to do is show that a dataset generated by a DAG also exhibits high CI. I’ve provided pretty graphs that demonstrate this is the case. I’ve provided a straightforward logical contradiction to the following:
phylogenies based upon true genealogical processes give high values of hierarchical structure, whereas subjective phylogenies that have only apparent hierarchical structure (like a phylogeny of cars, for example) give low values
I’ve done it at the gene level, and then reduced it to the nucleotide level and did the same.
I cannot think of anything I did incorrectly in all of this. If you have specific things you think I may have made a mistake in, please let me know, and I will do my best to correct it. I believe all my work is fairly easy to grasp with a bit of inspection, and I’m always willing to explain anything that doesn’t make sense.
The nested clades argument is primarily based on high phylogenetic signal in genetic datasets. I’ve shown genetic datasets can produce high phylogenetic signal, and have nothing whatsoever to do with common descent or evolution. As such, the nested clade argument must be said to be invalid at this point in time.
But, I understand that it is hard to accept something that flies against what the experts say. I currently have a post awaiting moderation over at theskepticalzone.com, where John Harshman, Joseph Felsenstein, and a variety of other evolutionary scientists hang out. We will see what they say.
I am sincerely trying to find some strong evidence of evolution which I can test myself. So far everything I’ve found falls apart when I dig into it. Probably you all suspect my motives or something, otherwise I’d think these holes I’m finding would attract more interest, or perhaps spur you to try and dig into the evidence more for yourself.
I don’t think that Talk.Origins has good updating. The consistency index has been largely abandoned because it is not very informative (actual evolutionary data sets give generally fairly similar results) and is influenced by multiple factors (such as how many taxa are involved). Although nested clades are a good measure of evolutionary consistency, CI is not that great of a measure of nested clades. But also, your examples of random data having high CI values are not addressing the question of nested clades and CI. Rather, what you need to look for is whether different trees generated for the same data show noticeable differences in the CI. If so, do the trees with high CI values show similar clades? Especially if you look at different data sets for the same taxa and consistently get higher CI for trees including certain clades and not others, that would suggest that certain clades are distinctly better supported and other groups are not. An evolutionary pattern would be expected to generate a reasonably consistent pattern of clades (though it would also be expected to produce some convergence, incomplete lineage sorting, etc.) Under non-evolutionary scenarios, there’s no reason for organisms that are similar in one way to be similar in other ways that do not link to the first. If the data in question do not reflect any evolutionary pattern, then different patterns with non-matching clades are likely to have near-equal support. The CI distribution for random data would be expected to be closer to a bell curve; the CI distribution for evolutionary data is expected to be more asymmetric across numerous tree configurations. As an added complication, how things like CI are calculated for a star-type tree varies. It’s true but trivial to say that a star tree does not generate any contradictions because it also does not give any information. If and how that insight is implemented in a particular evaluation program will vary considerably.
Thanks for this primer on the uses and misuses of Consistency Index analyses, David! The 2 key points I am deriving from your post are:
Biologists do not simply select the highest CI tree in a cladistic analysis and call it a day (which is, AFAICT, the approach @EricMH seems to have taken). Instead, they measure the stability of the clades in the vicinity of the CI peak, and only trust the outcome if that stability is found.
As the field has advanced, biologists are no longer relying on low-taxa CI analyses.
Have I grasped your key points correctly?
Noob questions:
What statistical metric would you use to measure the stability of cladograms in the vicinity of the CI peak?
You mentioned that “CI is not that great of a measure of nested clades.” What would be a better measure?
Also, if you could introduce a little more about yourself, I’d be grateful.
It’s always tough when some random guy on the internet comes in guns ablazing falsifying common ancestry.
It is curious how you have that special power when it comes to certain topics.
Well you probably do reject common ancestry because of very strong religious beliefs. Probably to the point where I would say your prior belief might override your ability to evaluate any evidence. E.g.
P(A|B) = 1 when the prior probability of A is 100% given any further evidence you could ever look at.
My personal opinion is that a more useful approach is not for you to try and falsify common ancestry, but to make specific predictions of what we should find given your model. What is your model? What sorts of predictions does it make of what we should find in nature? How well does it describe the evidence? Because a big mistake a lot of people make is “if I can just prove evolution wrong or silly, then logic dictates what I believe about the origin of species must be right!” Sometimes Christians use this same argument to prove completely different models which is just nonsense.
Sounds like I’m not the one making strong assumptions
Not the case whatsoever. The only reason I’ve turned away from evolution in the first place is ID. Before then I was happily on the way to rejecting my childhood Christianity and embracing atheism. Now that ID has convinced me evolution theory may not be as solid as claimed, and I’ve got a little knowledge in the matter, I’ve decided to try and verify evolution for myself, insofar as I can.
As such, knocking down individual pieces of evidence is exactly what I am doing. I have no idea what is an alternative theory, but I don’t believe there’s any need to have an alternative to show the status quo is faulty. And really, what I want is not to disprove evolution, but to find some really solid piece of evidence for evolution. Like as a programmer, if I’m trying to understand the behavior of a new language construct or library, I want to have something solid and repeatable to build my understanding. Additionally, as a programmer, when trying to troubleshoot a bug, I will create a hypothesis, and then try to eliminate the hypothesis with tests. I don’t need to have an alternate hypothesis besides “I don’t know” to debug my code in this way. The only thing worse that “I don’t know” is an incorrect hypothesis, so it is foolish to cling to an incorrect hypothesis just because I don’t have a correct hypothesis.
I’ve yet to find such a thing for evolution, and I’ve been working away pretty dedicatedly over the past year or so. It is strange that such a well established scientific field is hard for a lay person to validate, since the means are readily available with all the genetic data. And it is also strange evolutionary scientists are so allergic to questioning the fundamentals of the field.