Ha! Exactly what I did at first, at the gene level, and showed DAGs produce distinct trees just like genealogical processes do. The feedback was I needed to do this at the nucleotide level with official tools.
Now, I’m not very familiar with the PAUP software, but I think this addresses what you want. I create 100 random trees, run a likelihood test and sort the trees. If the clade structure is the same, then the sorted range should be pretty small, and visa versa.
There is a large DAG dataset that has 26 taxa.
There is a smaller DAG dataset with 18 taxa.
For comparison I have a primate dataset, with 12 taxa.
For the primate dataset I have a high score of 7300 and low score of 6394. For the 26 taxa DAG dataset I have a high score of 6768 and low score of 5857. For the 18 taxa DAG dataset I have a high score of 7816 and low score of 6915. The spread is the same in each case, but the larger DAG dataset has a lower overall score, which is to be expected with more taxa. So, there is not a clear difference between the clade structure in real world data and in the synthetic data, at least with regard to CI. I echo @Chris_Falter’s question, is there a different metric you recommend in place of CI? Hopefully a simple one ![]()
Reference results from primate data:
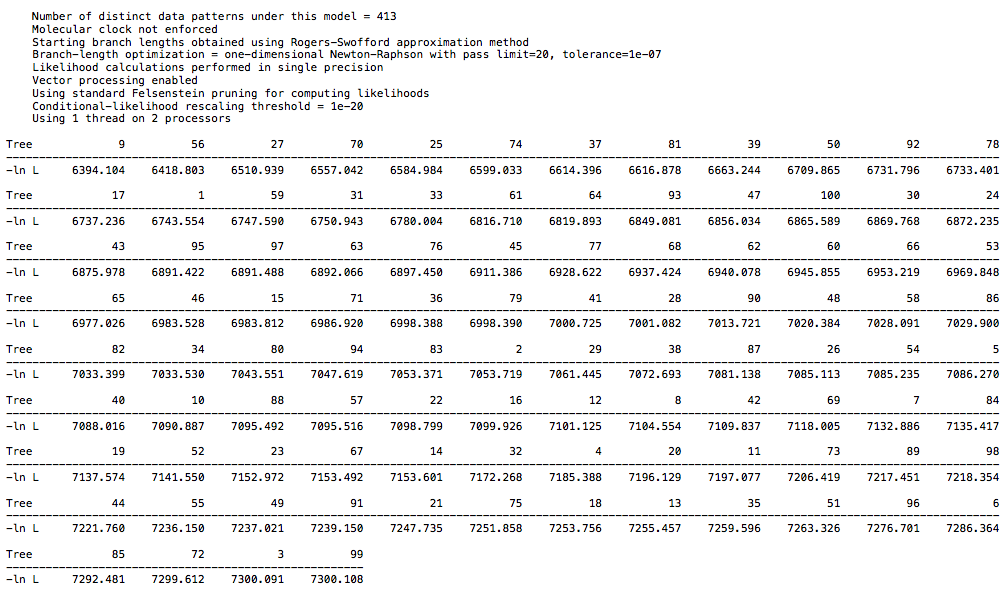
and from the 26 taxa DAG data:
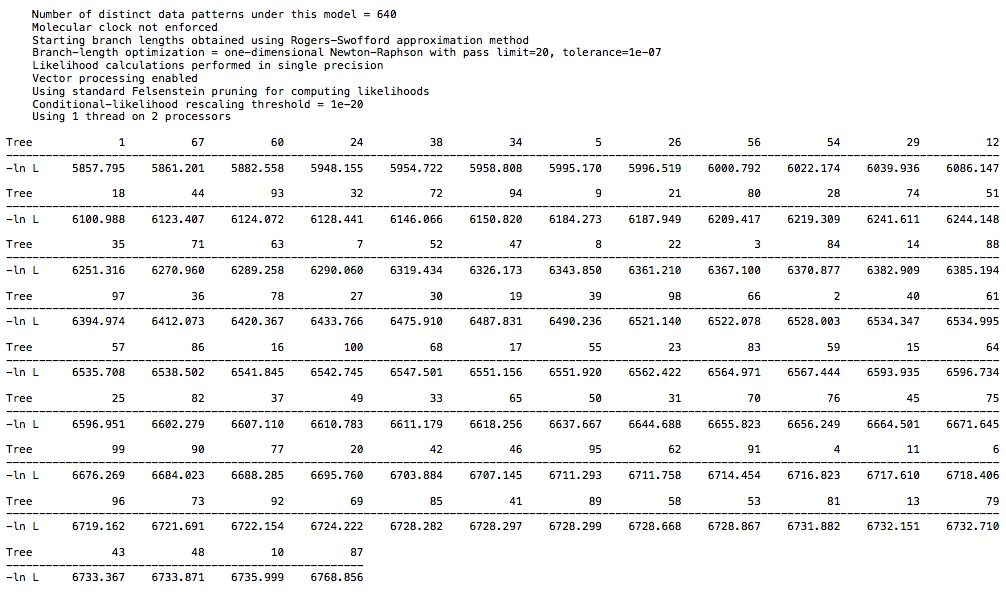
and from the 18 taxa DAG data:
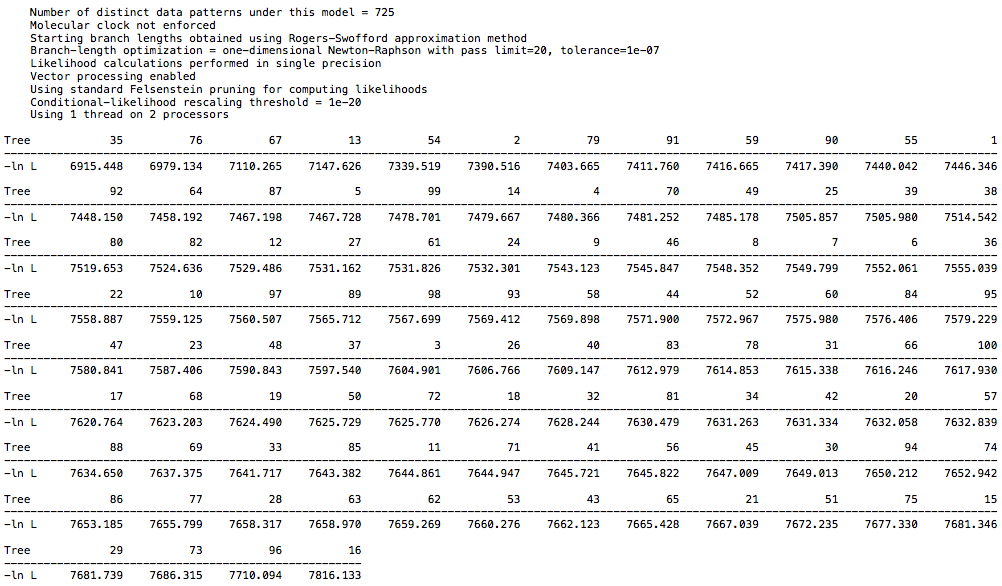
And let me know if you want the datasets to run the analysis yourself.