T_aquaticus
(The Friendly Neighborhood Atheist)
41
The first question would be to ask why this would result in ERVs that look like they evolved at many different levels of evidence, such as species distribution and LTR divergence. For example:
Why would a designer need to give humans an ERV at a specific position in their genome if that ERV is found at the same position between chimps and orangutans?
If a specific family of ERVs is found in the chimp and gorilla genomes but not in the human genome, why would a designer be forced to put the ERVs for that retrovirus family at different locations in the chimp and gorilla genomes?
Why would there be more divergence in the LTRs of a specific ERV if it is shared by many primate species than if it is shared by just two similar primate species?
Common ancestry and evolution explains all of these patterns of divergence and shared ERVs. If “original design” can not also explain these patterns then it isn’t a valid scientific explanation. On the flip side, could God create species so that they look like they evolved for no other purpose than to fool us? I suppose, but that doesn’t seem like a road that most theists want to go down. Therefore, if the evidence is consistent with evolutionary mechanisms and common ancestry and “design” can’t explain these patterns, then design is not the best explanation.
1 Like
gbrooks9
(George Brooks, TE (E.volutionary T.heist OR P.rovidentialist))
42
good questions. lets check if we can explain them under the design scenario:
since we already know that all apes are very similar to each other why not? its like any regular gene.
one possible explanation is that these ervs are indeed the result of viral infections. and b) i gave above at least 6 cases wich contradict this point, since they are in the same spot in different lineages.
im not sure what do you mean by that .are you talking about the molecular clock? (far species are more different from closer species in their ervs sequence).
as we can see: the design scenario can explain these points. on the other hand: how evolution can explain how some creatures that cant survive without these ervs existed before they got these ervs?
But I think @T_aquaticus is asking, then why exact locations in humans too? Surely we can reason, well chimps and orangutans sure, very similar species and common ancestor and thus highly shared DNA. But then why do humans have them in the exact same spots? Not just a few dozen but literally 6-8% of the genome comes from these shared ERVs!
No you didn’t. The Yohn et. al. 2005 paper are not endogenous retroviruses. They are exogenous retroviruses which leave a very different signature that is outlined quite extensively in the paper. Please review the difference between the two again. None of the 200,000+ endogenous retroviruses show a similar signature to the 6 you mentioned.
No. You haven’t demonstrated this at all. I mean again with the six exogenous retroviruses you keep mentioning- it was a retrovirus that infected two lineages independently (after they would have diverged in terms of their populations or were created if you believe chimpanzees and gorillas do not have a common ancestor). How is that anything related to an intelligent designer permitting the chimpanzee and gorilla lineage to become infected in these six spots but not the human and orangutan lineages?
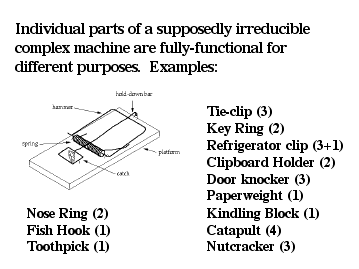
Outrigger, I really want to believe you are reading the posts and comprehending the things that are being posted (some of it is pretty complex stuff that challenges me to learn more). I believe you are trying to argue that “if some ERVs are functional, then how did species survive before the ERV insertion into their genome?” This was discussed above but it highlights one of the very creative aspects of evolution-that is the re-purposing of old material. Like Behe’s infamous irreducibly complex molecular motor, as the Dover Trial highlighted is simply just re-purposed. Thus, while some ERVs may have a function today, they most likely did not serve the same function millions of years ago. Much like how this mousetrap can work:
That question let’s me know that we are not on the same page about what endogenous retroviruses (ERVs) are and what the evidence actually is for common descent. Especially given that homo sapiens, nor even chimpanzees certainly were not ‘around’ 10-15 million years ago.
T_aquaticus
(The Friendly Neighborhood Atheist)
45
Then why don’t we see PtERV insertions at the same positions in the chimp and gorilla genomes?
The theory of evolution is able to predict which families of ERVs should be at the same position and why others should not, but I don’t see how Design can make any predictions. A designer could decide to you use completely different ERVs for chimps and humans. There is no reason we should see this pattern and only this pattern in the case of Design.
I am unaware of any publication that found an unambiguous example of an orthologous PtERV insertion shared by chimps and gorillas out of the hundreds found in each genome.
I am talking about the LTR sequences within one ERV in a single species. The LTRs are like the bookends of the viral genome, and they are identical to one another at the time of insertion. However, when we look at an ERV we see that the LTRs have diverged over time. The longer the ERV has been in the host genome the further the LTRs will diverge from one another as they accumulate different mutations. The theory of evolution predicts that an ERV shared by many primate species has been in primate genomes for a much longer time than ERVs shared by just two or three closely related species. Therefore, there should be more LTR divergence in an ERV that is shared by many primate species, and that is exactly what we see.
So how does design explain this?
2 Likes
T_aquaticus
(The Friendly Neighborhood Atheist)
46
Notice that it says "IF these sites were truly orthologous . . . ". They were unable to show that any of them were orthologous at 1 base resolution. From the paper:
BAC-based end sequencing is where you have large chunks of DNA (several hundred thousand base pairs) and sequence the very ends to determine where the large chunk fits into the larger genome. All they were able to determine is that the ERV was somewhere in those hundreds of thousands of bases. They were not able to determine where the ERV was down to the single base which is what you need for an unambiguous orthologous ERV.
For a few they were able to use published sequence to determine if they were at the same base.
“For the three intervals putatively shared between macaque and chimpanzee, we attempted to refine the precise position of the insertions by taking advantage of the available whole-genome shotgun sequences for these two genomes. For each of the three loci, we mapped the precise insertion site in the chimpanzee and then examined the corresponding site in macaque (http://www.ncbi.nlm.nih.gov). In one case, we were unable to refine the map interval owing to the presence of repetitive rich sequences within the interval. In two cases, we were able to refine the map location to single basepair resolution (Figures S4 and S5). Based on this analysis, we determined that the sites were not orthologous between chimpanzee and macaque. It is interesting to note that this level of refined mapping in chimpanzee revealed 4- to 5-bp AT-rich target site duplications in both cases. These findings are consistent with an exogenous retrovirus source since proviral integrations typically target AT-rich DNA ranging from 4 to 6 bp in length [24]. Although the status of the remaining overlapping sites is unknown, these data resolve four additional sites as independent insertion events and suggest that the remainder may similarly be non-orthologous. This apparent independent clustering of retroviral insertions at similar locations may be a consequence of preferential integration bias or the effect of selection pressure against gene regions, limiting the number of effective sites that are tolerated for fixation.”
same reference as above
gbrooks9
(George Brooks, TE (E.volutionary T.heist OR P.rovidentialist))
47
Gosh, @T_aquaticus, I have no idea what this sentence means… I’ve been holding my tongue for a day or two… and I just gotta know what this is supposed to mean - - “… asking for a friend!”
Why do you think they are in “the same spot”? That’s not what the Yohn et al. paper says. In fact, they look harder at a few of the similar insertions and conclude that they are NOT in identical spots, and thus are non-orthologous. I think you have misread Yohn et al., significantly.
Note that Yohn et al. was published 13 years ago, before full genome sequences were available for the various genomes. Not only did they not find insertions at “the same spot”, they didn’t have sequence data to make that determination for all of the similar insertion locations.
2 Likes
T_aquaticus
(The Friendly Neighborhood Atheist)
49
They looked at published chimp and macaque sequences and were able to determine the exact base where the ERVs started and ended in the genomes.
The method they used was BAC end sequencing. This is where you break up the genome into big chunks (~50k to 300k base pairs) and put them into bacterial artificial chromosomes (hence BAC) which are replicated in E. coli. They used a DNA or RNA probe to determine which of these big chunks of DNA had an ERV somewhere in them. They then sequenced about 1,000 base pairs of each end of the big chunk of DNA which allows them to figure out where the big chunk fits into the larger genome, but it isn’t able to tell them where the ERV is within that big chunk of DNA.
To use an analogy, we could treat an ERV like a street address. To determine if two people live in the same house you have to determine if they have the same exact address. The BAC end sequencing method can only tell you which town they live in, but it can’t tell you what street they live in or what their house number is. If there is published sequence for the region where the ERV actually inserted, then you can get the street and house number and determine if two ERVs live in the same house.
T_aquaticus
(The Friendly Neighborhood Atheist)
50
This misreading of the Yohn et al. paper is one put forward by “professional” YEC/OECs and ID supporters. The article at ENV is a good example:
It is the same tune that we have seen for a while. Well meaning Christians go to these sites, trust them, and then start spreading the falsehoods found on those pages. I have even shown Discovery Institute staff the factual errors on those pages, but there it remains. From the article:
“Out of tens of thousands of ERV elements in the human genome, roughly how many are known to occupy the same sites in humans and chimpanzees? According to this Talk-Origins article, at least seven. Let’s call it less than a dozen. Given the sheer number of these retroviruses in our genome (literally tens of thousands), and accounting for the evidence of integration preferences and site biases which I have documented above, what are the odds of finding a handful of ERV elements which have independently inserted themselves into the same locus?”
This was written in 2011, 6 years after the chimp genome paper demonstrated that humans and chimps share more than 99% of their 200,000 ERVs.
these specific ptervs are indeed the result of viral insertions.
these ervs are species specific.
basically lts like any other gene. we already know that a chimp and human for instance are more similar to each other since they are also similar in their morphology. therefore we can predict even under the design scenario that as far as we are talking about far species- their ervs should be more different too and vice versa (at least in most cases).
two points:
even if we are talking about 300 kb region- out of 3 billion bp genome its about 1\10000 of the genome. this is why i call it a “specific spot”. and not for free they are claiming for “preferential integration bias”.
by this criteria any orthologous gene between chimp and human isnt a real orthologous since there is about 2% difference between them.
T_aquaticus
(The Friendly Neighborhood Atheist)
52
Why would species specific ERVs fall into the same phylogeny predicted by the theory of evolution? Why would a designer be limited to what evolution would do?
That doesn’t make any sense. There is no reason that a designer would have to make species similar to each other to begin with. There is no reason that a designer would have to use similar ERVs for similar looking species. On top of that, there is no reason that a designer would have to fit species into a nested hierarchy. When humans design organisms we take mix and match genes from very different organisms and combine them into another organism. For example, we have taken a green fluorescent protein from jellyfish and put an exact copy of that gene into mice. So why would design predict a nested hierarchy when designers notoriously do not fit their designs into a nested hierarchy?
On top of that, I am talking about the LTRs of a single ERV in a single species. The viral genome looks something like this:
The LTRs are like bookends on the viral genome. When the virus is copied and inserted into the genome those LTRs have identical sequences at either end of the genome. When I talk about LTR divergence I am talking about differences between the LTR on the left and the LTR on the right in a single species and in a single ERV, not the divergence between LTRs in different species.
How does Design explain how ERVs shared by many primate species have more differences between the left and right LTR in a single ERV than in an ERV shared by just 2 closely related species?
When we say that a chimp and human ERV are orthologous we are not saying that they are within 300,000 bases of each other. We are saying that the insertion is at the same orthologous base. Therefore, if you are going to claim that there are orthologous ERVs that contradict the expected pattern then you need to show that they are at the same base.
That’s not how orthology works. The sequence of an ERV does not determine its location in the genome.
4 Likes
gbrooks9
(George Brooks, TE (E.volutionary T.heist OR P.rovidentialist))
53
What does THIS particular sentence mean?
T_aquaticus
(The Friendly Neighborhood Atheist)
54
They determined that the ERV insertions were at different positions in each genome. To use the analogy, they live in different houses.
thanks but i actually referring to the question of how the creature survived before it got the viral parts. a rat for instance cant survive without a specific erv called syncytin. so if we go back in time before the rat got this gene, how the rat survived?
1 Like
gbrooks9
(George Brooks, TE (E.volutionary T.heist OR P.rovidentialist))
56
Ahhhh, @outrigger, now THAT is interesting! I had never heard of syncytin! I look forward to @T_aquaticus or @pevaquark answering your question!
T_aquaticus
(The Friendly Neighborhood Atheist)
57
There are tons of species that survive without that gene, so it isn’t required for survival for all species. Genes can be advantageous at first, but not required. Over time the species evolves other features that depend on that new trait so that it becomes required over time. This is known as the Mullerian Two-Step:
“… thus a complicated machine was gradually built up whose effective working was dependent upon the interlocking action of very numerous different elementary parts or factors, and many of the characters and factors which, when new, were originally merely an asset finally became necessary because other necessary characters and factors had subsequently become changed so as to be dependent on the former. It must result, in consequence, that a dropping out of, or even a slight change in any one of these parts is very likely to disturb fatally the whole machinery; for this reason we should expect very many, if not most, mutations to result in lethal factors …”
Muller, H. J. (1918) “Genetic variability, twin hybrids and constant hybrids, in a case of balanced lethal factors.” Genetics 3:422-499.
first; i dont think that evolution predict this. some scientists actually think that chimp isnt the closest species to human. secondly: id predict it too since we know that similar morphology= similar genetics in general. its just a simple observation.
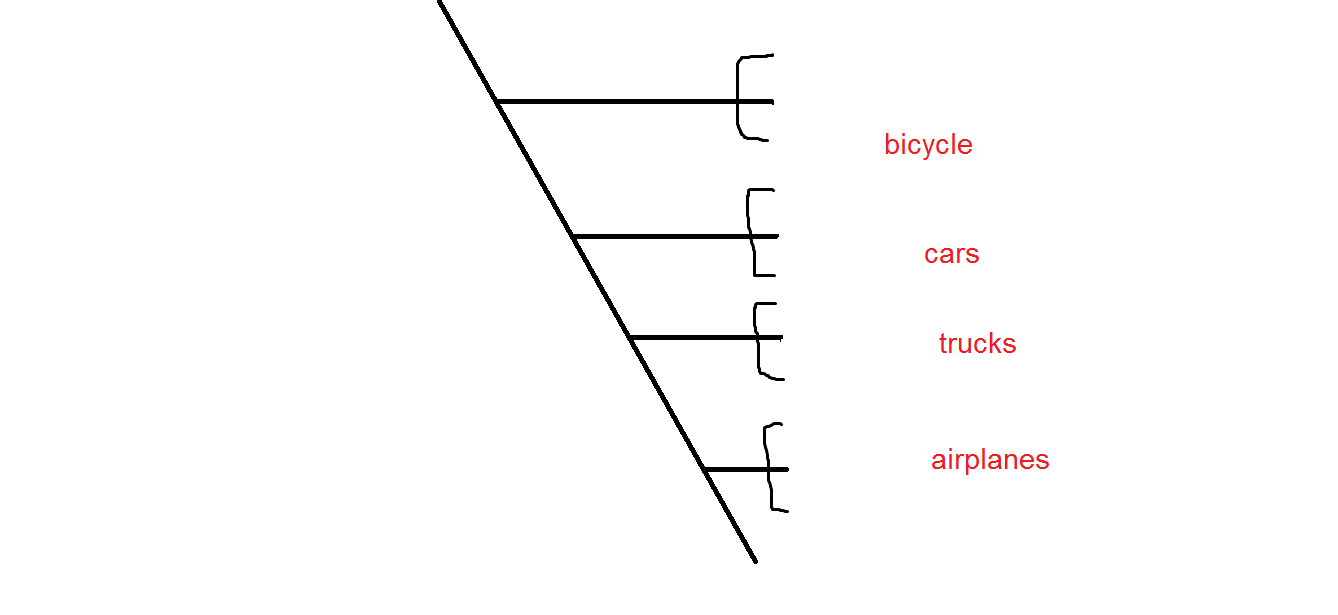
if so why so many vehicles are so similar to each other?
i think that they are actually do. see this for instance:
are you saying that human for instance has some ervs that are very different in their left and right LTR compare to other ervs LTR in the same genome? if so can you give an example?
but chimp and human are about 2% different. so how we can claim that their ervs are in the same base in the genome?
its only a theory. its like saying that since some cars can work without a fuel (an electric car) then it means that we can made a car stepwise.
gbrooks9
(George Brooks, TE (E.volutionary T.heist OR P.rovidentialist))
59
Maybe this is a more complete definition for the term “orthologous”:
“Orthologs are genes in different species that evolved from a common ancestral gene by speciation.”
“Normally, orthologs retain the same function in the course of evolution. Identification of orthologs is critical for reliable prediction of gene function in newly sequenced genomes. (See also Paralogs.).”
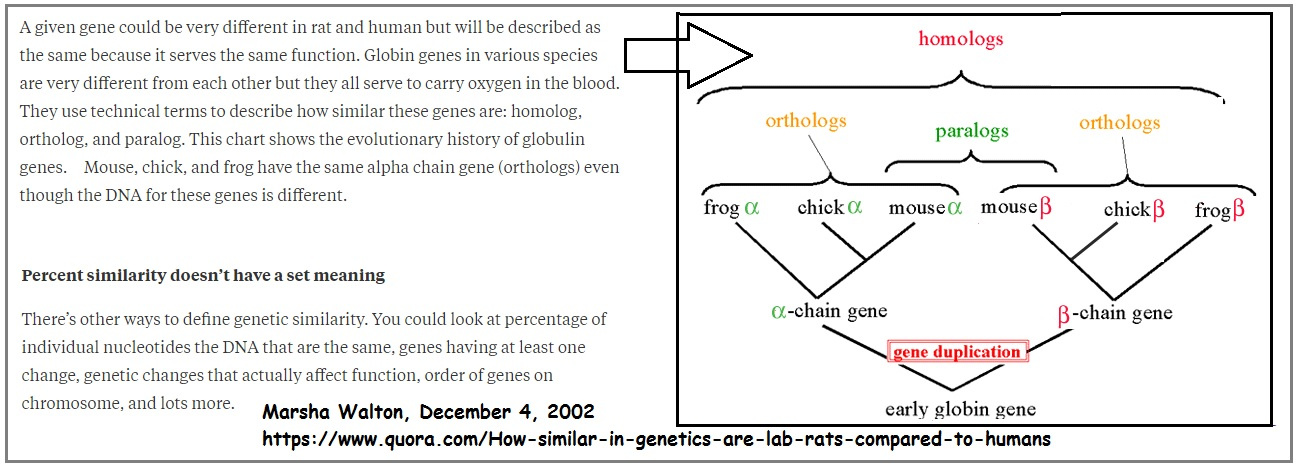
DEFINITION OF HOMOLOG, ORTHOLOG AND PARALOG
It was suggested that I might want to provide definitions which made the difference between these terms clear. These definitions are provided here.
Homolog
A gene related to a second gene by descent from a common ancestral DNA sequence. The term, homolog, may apply to the relationship between genes separated by the event of speciation (see ortholog) or to the relationship betwen genes separated by the event of genetic duplication (see paralog).
Ortholog
Orthologs are genes in different species that evolved from a common ancestral gene by speciation. Normally, orthologs retain the same function in the course of evolution. Identification of orthologs is critical for reliable prediction of gene function in newly sequenced genomes. (See also Paralogs.).
Speciation
Speciation is the origin of a new species capable of making a living in a new way from the species from which it arose. As part of this process it has also aquired some barreir to genetic exchage with the parent species.
Paralog
Paralogs are genes related by duplication within a genome. Orthologs retain the same function in the course of evolution, whereas paralogs evolve new functions, even if these are related to the original one.
If I had two sentences how similar would you say these are:
Bob went to the store and bought some lemonade, soda, and bottled water.
Bob went the store and bought some lemonade, soda, and bottled water.
If you compared the content in order, you would say the 2nd sentence shares 12/13 words in the same order. However, if you just compare spot by spot the bottom sequence only shares 2/13 words in the same spot. Which of the two is a better comparison?
You have many base pairs if you lined up all 3 billion of ours and let’s say 3 billion of a chimpanzee, perfectly aligned in some common ancestor. And then two populations split and our has a base pair deletion close to the beginning. Would the two then, despite being over 98% similar all of a sudden be very very different?