T_aquaticus
(The Friendly Neighborhood Atheist)
101
That is absolutely false. Computational phylogenetics is able to test proposed evolutionary trees.
“Computational phylogenetics is the application of computational algorithms, methods, and programs to phylogenetic analyses. The goal is to assemble a phylogenetic tree representing a hypothesis about the evolutionary ancestry of a set of genes, species, or other taxa.” Computational Phylogenetics–Wiki
If you construct a tree and randomly disperse traits throughout the tree then the algorithms used in these tests will tell you that there is no statistically significant phylogenetic signal. These trees aren’t assumed. These trees are tested as a hypothesis, and they can be falsified.
That is wrong. Let me repeat. THAT IS WRONG.
If a mouse and a jellyfish shared a gene with 100% sequence similarity, and no other mammal or vertebrate had a gene with similar sequence, then this would falsify evolution.
Again, it is the PATTERN of similarity that evidences common ancestry and evolution, not simply similarity. That pattern can be tested, and it isn’t assumed. That pattern is a nested hierarchy, or otherwise known as a phylogenetic signal.
Can you tell us why this is a bad conclusion?
T_aquaticus
(The Friendly Neighborhood Atheist)
102
Only a tiny percentage of most eukaryotic genomes are functional, and only those parts can be affected by the mechanisms you have highlighted. Most of those genomes are non-functional and they change through genetic drift. We get the same phylogenies when we compare non-functional DNA which means that the arguments you are making fall flat on their face.
The reason I didn’t quote that was because I don’t hold to the world view that the only two options are common ancestry or separate ancestry.
The fact that common descent is more probable than separate descent (which it should be) does not make common descent true.(Besides, the fact that I shared the link should tell you that there is no intention to quote mine or pull a quick one).
And how does this change the fact that at least the author of the book admits that we can’t really prove hypotheses of common descent through genetics without the genome of related common ancestors?
These are all probability analysis based on an assumption of common descent. Its relative probability, which means that it says one species is closer than another in terms of having more similar genes in the area compared.
First off, it’s a very interesting paper.
As per the paper, 95.8% of the ERVs were found in “non-orthologous” regions…i.e they did not mess up any “functioning” genes and lead to diseases, that would cause these genes to be selected against and lost…
Other reasons are -
a) Faster than expected divergence (i.e some amount of novelty difficult to explain by plain mutation and inheritance has happened in the common ancestor).
b) The genes which “express” themselves transcript differently in humans and chimps (i.e no inheritance of function).Indicating some kind of co-option.
Ortholog genes usually denote genes with function which have been inherited. If common descent is true, this should be the vast majority of the human genome that has function.(because mutations leading to novelty in function is rare… and evolution is a step by step process).
There are papers which put the of the human genome with function and subject to natural selection between 8.2 to 10 percentage.
Others put the percentage at max 25 based on “mutational load”…
And of course ENCODE famously put the no: at 80%.
Another interesting feature is a recent study on the yeast cell (representing a billion plus years of “evolution”) which found that the percentage of genes with identifiable function in yeast is 90% (That’s a rise from 30%). They basically sequentially knocked out 3 gene pairs from the yeast cell, and measured for fitness variation… Turns out 90% has a function. http://rsob.royalsocietypublishing.org/content/7/6/160330
We should not get anything near that with human beings…
However, If they do find 80+% of the human genome has function, then we will have to find an answer to how come the ERVs ended up bonding to functional DNA and not get selected against…
And if functions are found for most of the ERVs, we will have to explain the probability of the right genetic code ending up in the right place in the right time so often…
I expect future findings to continue proving that the human genome has function… And that ERVs also have function…
So yes, a high percentage of function for the genome should falsify evolution. (At least I think so).
And it’s encouraging to see biologists conduct experiments to find empirical data about gene function…
T_aquaticus
(The Friendly Neighborhood Atheist)
104
An ERV insertion found at the same spot in the same gene in two different species would be an orthologous ERV. Orthology has nothing to do with function. It has to do with position in the genome.
All that means is that there was positive selection for mutations in those ERVs which caused them to diverge at a higher rate than predicted by neutral drift.
Humans don’t have PtERV insertions, so I’m not quite sure where you are getting that from.
Non-functional DNA can also be orthologous. Orthologous simply means being in the same position in each genome. It has nothing to do with function.
That’s because ENCODE uses a definition of function that simply means “it does something”. This is not the definition that other scientists are using. If a stretch of DNA is transcribed into RNA at least once in one cell, then ENCODE considers it to be function even if that RNA has no impact on the fitness of the organism. The vast majority of biologists define functional DNA as DNA sequence which has an impact on fitness and will therefore show some evidence of sequence conservation. ENCODE includes junk DNA in their definition of functional DNA because even junk DNA will be transcribed into RNA at low levels.
About 2-3% of the human genome is made up of genes.
You falsely assume that there is only one right sequence of DNA.
I’m not sure how exactly your yeast paper has anything to do with all the papers written on human beings where much less than 20% has any function in the sense of fitness.
But they have not been heading this way… for a whole decade. The only paper at all was the ENCODE project which is discussed in length above… certainly they had a very “liberal” definition of function that many non-experts jumped on.
Also function has nothing to do with the ERV evidence.
Not in the sense that any other study (or human being, really) uses the word ‘function’. The comparable number from ENCODE is around 11%.
That’s 90% of genes (protein-coding genes, specifically). I would expect at least 90% of human genes to have functions, too. But protein-coding genes make up less than 2% of the human genome.
Any activity involving creating phylogenetic trees involves a few basic assumptions. Whether its morphology, or computations of genetic similarity, the following assumptions are basic.
Three things assumed are -
Change in characteristics occurs in lineages over time.
Any group of organisms is related by descent from a common ancestor.
There is a bifurcating, or branching, pattern of lineage-splitting.
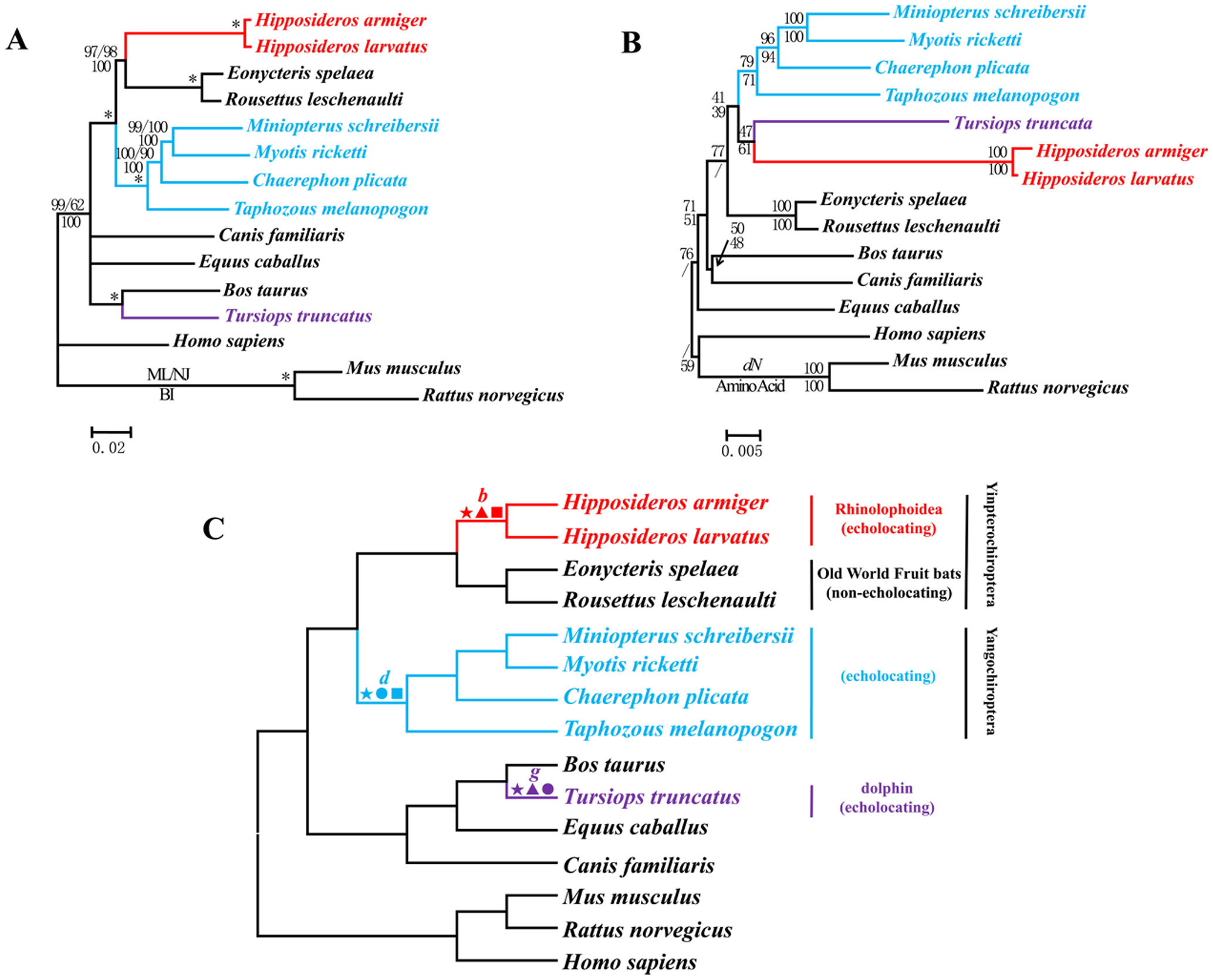
This is a misunderstanding on your part. For example, if you make phylogenetic trees from genes associated with echolocation between Bats and Dolphins, you would get some thing like this
As to phylogenetic signals. It involves removing genetic noise (genetic information that does not tell us anything about lineage) and thus is a filtered result in itself.
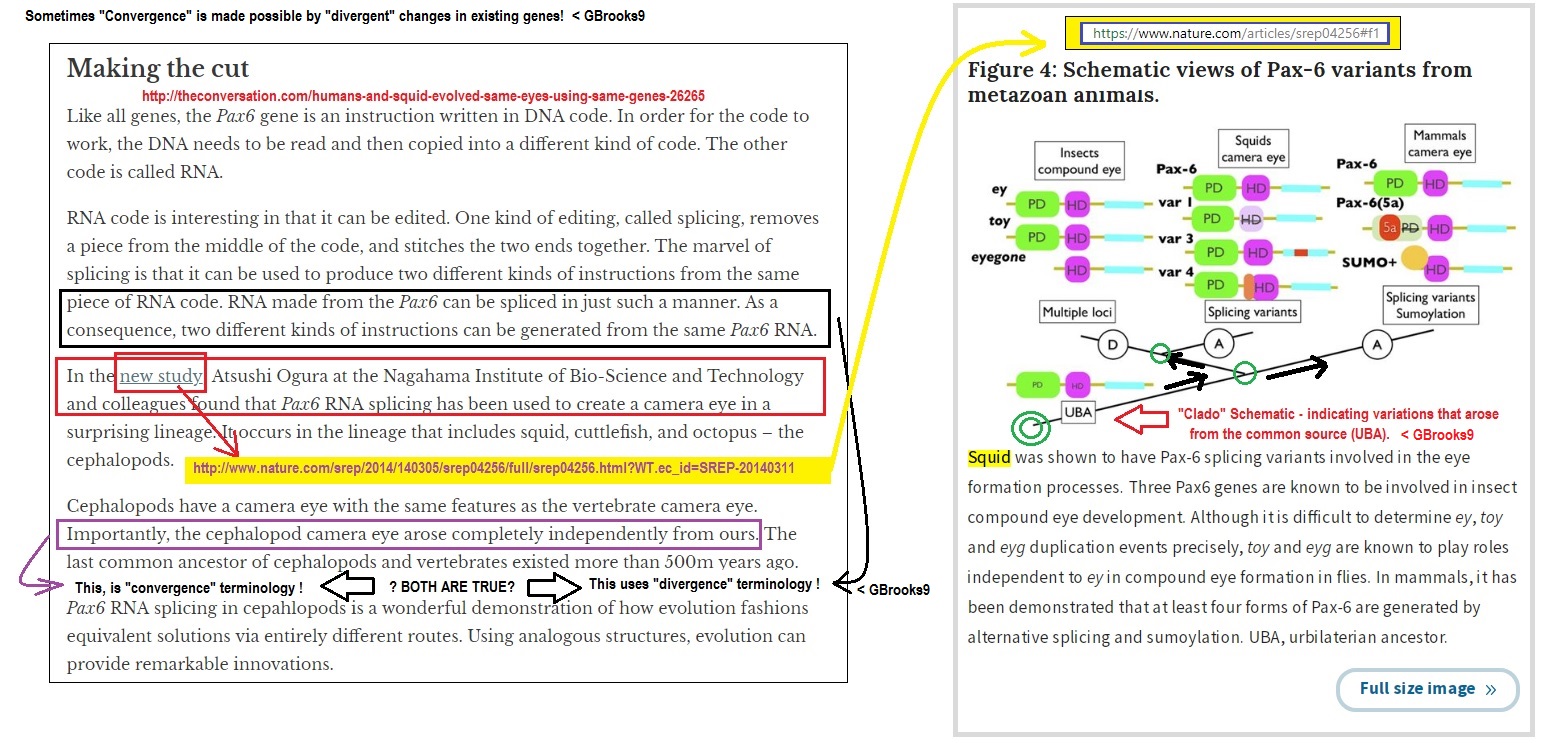
Anyway, with respect to your comment on jelly fish and mice… would Squids and human beings do?
As is well known, Squids and human beings have eyes of the same type. Recent studies have shown that the development of the camera eye in Humans and squids happen through the same gene called ipax6. The differences in ipax gene for squid which allow the squid to develop camera eyes are not shared with other molluscs and so this is assumed to have evolved “convergently”. https://www.nature.com/articles/srep04256?WT.ec_id=SREP-20140311
Blockquote
In summary, we identified the acquisition of splicing variations of Pax-6 in cephalopod eyes (Figure 4) and found that the acquisition occurred independently in vertebrates and cephalopods. These Pax-6 splicing variations in cephalopods were controlled spatio-temporally during eye formation. Although the acquisition of camera eyes in the cephalopods is yet a problem, Pax-6 variants in cephalopods have been acquired in a lineage-specific manner.
Blockquote
Of course, now evolutionary biologists are speculating that a common ancestor (of the octopus and humans) had these genes and they were lost. I am going to quote from an article below which argues for “constrained genes” in octopus-
Blockquote
In spite of the evolutionary divergence between octopuses and humans, 69.3% of the genes examined (729 of the 1052 genes) were commonly expressed in the camera eyes of human and octopus. Moreover, comparison of octopus-eye ESTs with genes in the human connective tissue indicates that the similarity of gene expression between human and octopus eyes should be remarkable. Note that the increase of gene expression similarities
between human and octopus eyes from 15% (162/1052) to 69.3% (729/1052) is caused by the increase of the EST data set of human eye from only 3809 ESTs in the database of BodyMap to 13,303 human-eye ESTs in the combined database of NEIbank, MGC, and BodyMap. This observation suggests that many more similarities of gene expression between human and octopus eyes will be observed when the EST data increase further.
Although these 729 genes might contain housekeeping genes because the 44 genes were also found in the ESTs of human connective tissues in BodyMap, we found that many more
genes (118 genes) were specific to the camera eye in this case. Therefore, we suggest that these 729 genes contain genes necessary for the developmental process and biological function of the camera eye.
Blockquote
For the evolutionary origin of the gene set working for the camera eye, we observed that 1019 genes existed in the genome of the common ancestor of the bilaterian animals. Although the morphology of the ancestral eye cannot be inferred from this study,
we were able to provide strong support for the hypothesis that these genes having had an important role in the function of camera eyes in both humans and octopuses were present in the last common ancestor of these two lineages.
Blockquote
Blockquote
We found that some of
the genes conserved between humans and octopuses have been lost in the organisms having no camera eye structures.This result supports a hypothesis that the genes required for the development and maintenance of the camera eye were retained in the vertebrate and octopus lineages, but lost in insects,nematodes, and tunicates. For insects, it is likely that the
genes having functions specific to the camera eye were not important for the evolution of the compound eye, and therefore must have been lost from the genome.
Blockquote https://pdfs.semanticscholar.org/9f21/a4dc71304d8f7cd95ec625fb7a20adcce4e2.pdf
So the answer to your Jelly Fish, mouse claim is simple. There are three possible evolutionary scenarios -
They both gained similar genes through parallel evolution/convergent evolution.
There was a common ancestor with these genes. These genes were conserved in the two lineages and lost in everything else (what a relief there are no actual common ancestors of intermediate species to actually verify all this!)
Any combination of 1 and 2 which either works or leaves people so confused, they dont know which is which.
Well, first of all we do not see such a clear relationship between similarity and common descent.
refer discussion on development of the eye given above.
Blockquote
Despite the pressing need to identify and characterize all functional elements in the human genome, it is important to recognize that there is no universal definition of what constitutes function, nor is there agreement on what sets the boundaries of an element. Both scientists and nonscientists have an intuitive definition of function, but each scientific discipline relies primarily on different lines of evidence indicative of function. Geneticists, evolutionary biologists, and molecular biologists apply distinct approaches, evaluating different and complementary lines of evidence. The genetic approach evaluates the phenotypic consequences of perturbations, the evolutionary approach quantifies selective constraint, and the biochemical approach measures evidence of molecular activity. All three approaches can be highly informative of the biological relevance of a genomic segment and groups of elements identified by each approach are often quantitatively enriched for each other. However, the methods vary considerably with respect to the specific elements they predict and the extent of the human genome annotated by each
Blockquote http://www.pnas.org/content/111/17/6131
The evolutionary view of function as something on which selection acts upon is a tautology.I dont see it as either valid or particularly valuable.
Anyway, the important thing is that the NIH which funded ENCODE is looking at it from a prevention/cure of disease point of view and so they should do a more thorough empirical search than any evolutionary biologist.
Hence my optimism.
Its an example of empirical studies vastly increasing our knowledge of function in genes.
There are similar studied in progress on the human genome also which should push the envelope on “function”
Perhaps… i am not sure though. The paper keeps claiming that their study is “genome wide”… (I have given the link for the paper,you can verify it yourself).
Also recently, John Hopkins started a project to create a synthetic yeast cell. They claim the yeast cell is 8% smaller than the natural yeast cell after removing “junk DNA”
Blockquote
One exciting feature of the synthetic genome is that it’s about 8 percent smaller than the natural yeast gnome. Genetic sequences that can make DNA disposed to mutations and instability were relocated and noncoding “junk” DNA was removed.
Blockquote https://www.biosciencetechnology.com/news/2017/03/synthesizing-yeast-genome
So these guys obviously felt some need to keep 92% of the genome.
However, i just pointed to the paper as a example of how concerted empirical/laboratory work can vastly increase the percentage of the genome with known functions.
gbrooks9
(George Brooks, TE (E.volutionary T.heist OR P.rovidentialist))
110
The gene cited has a common ancestor… but that gene’s sequence for humans is NOT the same as the sequence for that gene used in Squid!
In fact, it suggests that there was a common population for use of the original version of master controller gene… and that the squid version sent squid eye in a direction different from how fish and human eyes developed.
Can you cite any paper that shows how different the gene is?
Having a common ancestor means the genes show homology… I.e they are similar.
I suggest you read the second paper I cited, or at least the part I quoted.
I don’t know where you got this from. Even the article you linked to doesn’t say anything of the sort.
Let me repasts the quote from the paper I cited.
I couldn’t find any document saying pax6 genes of humans are more similar tot hat of fish as opposed to cephalopods… is that what you are claiming? Can you share your source for the info?
gbrooks9
(George Brooks, TE (E.volutionary T.heist OR P.rovidentialist))
112
The reason you don’t recognize the signs that genetics are diverging is because you
only notice the similarities being described, rather than the differences that emerge in
an “ancestral” control gene, in order to make what you think are “the same” eye.
Yes, both eyes are “camera box”, but that’s just one level of the nested hierarchy.
Labels in nested hierarchies are not arbitrary or tautological, because the labels are
based on Actual Differences in the biological structure. You are right that human language
can be rather arbitrary - - but you incorrectly attempt to make the arbitrariness of language,
and of the humanly subjective selections made by an investigator, as equally arbitrary.
If there are REAL differences, then the point of the nested hierarchy is to track the changes,
where they might have come from, and the pattern of where a change has ended up.
[[ Be sure to click on the image to maximize font size! ]]
[[ Be sure to click on the image to maximize font size! ]]
T_aquaticus
(The Friendly Neighborhood Atheist)
113
Computational phylogenetics tests those assumptions. If those assumptions were not correct then you wouldn’t get a phylogenetic signal.
When you compare the whole Prestin gene you get the expected phylogeny:
“Repeated analyses of phylogenetic reconstruction based on nucleotides corresponding to variable amino acid sites only (414 bp), thus reflecting areas of nonsynonymous change, also recovered the putative gene tree topology with strong support for the clade of laryngeal echolocators (62% ML bootstrap and 97% BPP). Conversely, analyses of the remaining nucleotides (1,800 bp) recovered the species tree, albeit with reduced support for the Yinpterochiroptera clade (<50% ML bootstrap, 47% BPP). Therefore, the monophyly of laryngeal echolocators is supported when only those parts of the gene that lead to amino acid changes are analyzed, but this arrangement is lost and laryngeal echolocators become paraphyletic (as suggested by recent molecular phylogenies) when areas of the gene that do not result in amino acid changes are analyzed.” http://www.pnas.org/content/105/37/13959
What gene do humans have that is an exact copy of a jellyfish gene that is also not found in any other vertebrate? That was the challenge. All other vertebrates have Pax6, and the sequences produce the expected phylogeny.
As to the structure of the eyes themselves, the cephalopod and vertebrate eyes are very different. The retinas in each develop from different cell types and the retinas face in opposite directions.
They are found in numerous vertebrate species which fails the challenge that was given.
T_aquaticus
(The Friendly Neighborhood Atheist)
114
An interesting aside . . .
ENCODE listed introns as being functional simply because they were transcribed into RNA. The authors of the paper below removed introns from yeast genes and observed that only a minority of those introns affected fitness in any meaningful way:
“Together, our results show that although the presence of introns may optimize gene expression and provide benefit under stress, a majority of introns could be removed with minor consequences on growth under laboratory conditions, supporting the view that many introns could be phased out of Saccharomyces cerevisiae without blocking cell growth.” Parentau, et al. (2008)
It is genome-wide – a genome-wide study of gene knockouts, as they state clearly.
Well, good. I don’t know what fraction of the yeast genome (ignoring the fact that “yeast” covers a wide range of species) is junk, but I would expect it to be smaller than for the human genome. Single-celled organisms are typically under more selection for rapid replication and minimal waste than something like humans.
Sure, but that’s irrelevant to changing the percentage of the genome that is known to be functional, which is a very different question. We already know that human genes are functional even when we don’t know what they do. We know because of conservation between species in genes, because of reduced diversity around genes, and lower derived allele frequencies in the functional parts of genes. For the same reason, we know that much of the genome doesn’t have function.
The problem is that the definition of “functional” adopted by ENCODE is utterly useless from a prevention/cure of disease point of view. The whole point of ENCODE was to give clues about which genetic variants were most likely to be affecting phenotype, and in particular to be affecting health (I was there – I know). We had increasingly many studies showing association between multiple variants in a chromosomal region and some physical trait, but little clue as to which variant actually caused the physical difference. ENCODE exists to give us lists of likely functional elements, to tell us which variants to examine first. Much of the content of the ENCODE studies is useful for this purpose. For example, knowing that some transcription factor binds at a site makes it more likely that that site is functional (although still not particularly likely – most of the putative binding sites seem not to be functionally important). Knowing that 80% of the genome is biochemically active – in most cases, meaning just that it’s transcribed some of the time – does almost nothing to help in this endeavor.
I’m fine with the idea that some bits of the genome could have an effect on phenotype without leaving an evolutionary trace (although I expect that to be pretty rare), but ENCODE’s definition of function is a lousy way to detect such sites.
Can I just publicly laud @pevaquark’s new jedi moderation skills?
I cracked. Up. When saw this new thread title. bahaha well done
2 Likes
T_aquaticus
(The Friendly Neighborhood Atheist)
118
How does separate ancestry fit the data as well as common descent? Why would life with separate origins use the same tRNA, or use tRNA at all?
Do you have that evidence?
We have fossils of small vertebrates from the Cambrian who didn’t even have bones or a skeleton of any kind:
The fossils of that species are 525 million years old. If those fossils can survive to the present day I don’t see why a fossil of a big, bony mammal could not.
1 Like
gbrooks9
(George Brooks, TE (E.volutionary T.heist OR P.rovidentialist))
119
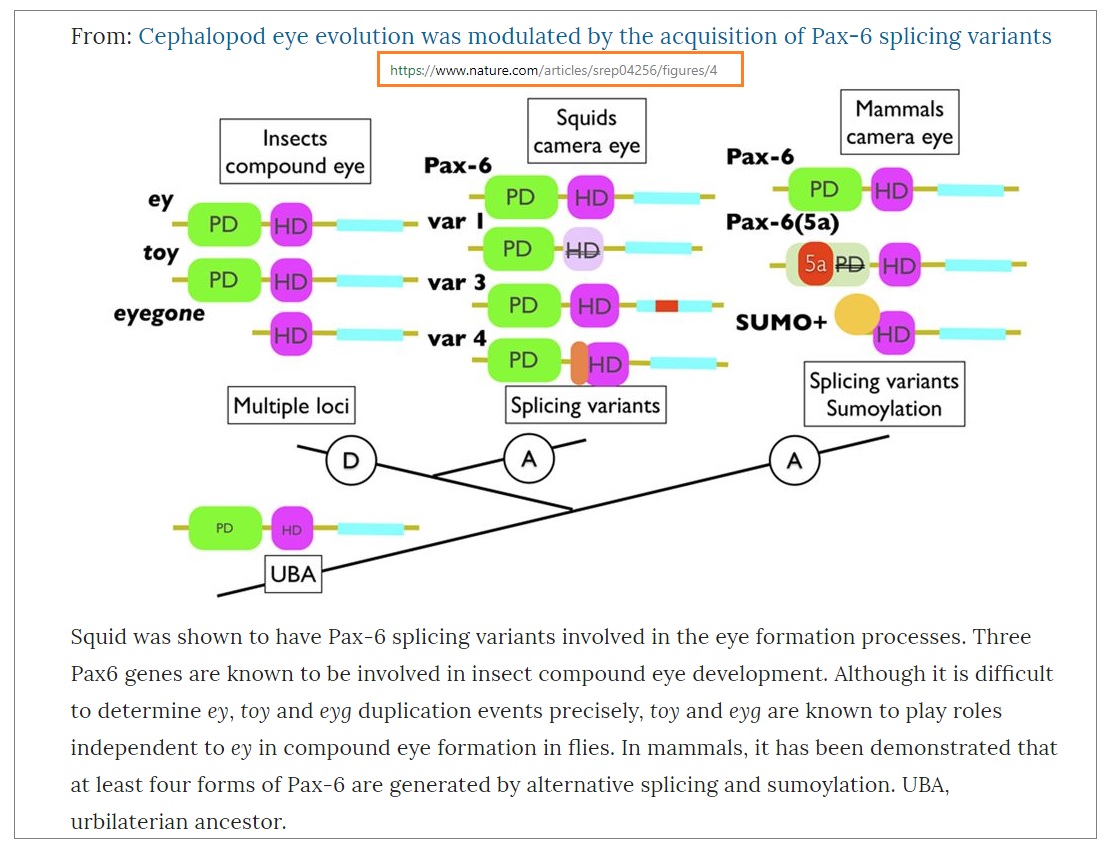
Think the most dramatic differences are between the specific configuration of a gene known
as Pax-6:
Are found in a three-way compare/contrast:
Insect Compound Eyes (Invert) - Multiple Loci: ‘ey’, ‘toy’ and ‘eyegone’ variants
Squid Camera Eye with no Blind Spot (Invert) - Splicing Variants: ‘Var1’, ‘Var3’, ‘Var4’
Human Camera Eye with Blind Spot (Vertebrate) - Splicing Variants/Sumoylation: Pax-6, Pax-6(5a), SUMO+
The thing to note from these results is that Scientists can name a gene based on its function, as in
Pax-6, without necessarily saying that the genes are identical. Somewhere in this gene is information for
how to splice the proteins … and this varies with the branch of life under consideration.
You have rather cavalierly assumed that when a headline says “same gene” and “same eyes”, that this
is literally true. And this journal article proves that it isn’t. The gene differs by something as subtle as a
splicing logic. And the eyes differ in terms of blind spot (squid do not have one), as well as sensitivity to
low levels of light vs. competent functioning in extreme lighting conditions in which humans have to function (as various as the inside of a cave and the outside of a cave hunting in the bright sun). There are also
differences in geometry and tissues that further accommodate differences that are important to Squid more
than to humans.
I dare say that virtually any “topic” you might bring to bear on your crusade (like echo location) suffer from
the same over-generalizations, and presumptions that would be irrelevant to any competent lab worker.
T_aquaticus
(The Friendly Neighborhood Atheist)
120
I’m not sure if the following link will work, but it is a link to the pairwise alignment scores for Pax6 amongst model organisms:
As we would expect, sequence similarity correlates with evolutionary distance. There is also the expected pattern of genetic equidistance:
Pax6
Human v. mouse = 94.3% similarity
Human v. chicken = 87.5% similarity
Mouse v. chicken = 87.2% similarity
Evolution predicts the evolutionary distance between mice and chickens is the same as the evolutionary distance between humans and chickens, and that is supported by the % similarity between the genes in each species. IOW, the common ancestor for mice and chickens is the same as the common ancestor for humans and chickens. That is what we see in these numbers.