Hi all,
To explore further the effect of improvement of the chimpanzee genome assembly on percentage similarity estimates for entire human and chimpanzee genomes, I have done the same calculation for the PanTro4 genome assembly (based on 6X Sanger read coverage) as I did earlier for the PanTro6 genome assembly (based on a lot more sequence data and covering more of the chimp genome - see my previous post).
Here are the two sets of stats side by side:
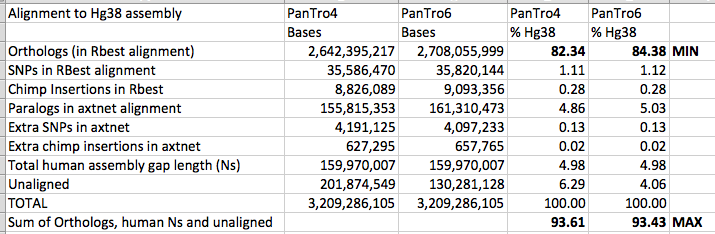
The improved chimpanzee genome has led to slightly greater precision in my estimates of human chimpanzee percentage similarity: the minimum is raised, and the maximum is slightly reduced.
One thing that I was not necessarily expecting is that the size of copy number variant (CNV) regions seem to have increased in size with the improved chimpanzee genome assembly (as shown by the “Paralogs in axtnet alignment” row). I had wondered if improved chimpanzee coverage might decrease this figure, as repeats in the chimpanzee became better resolved, but this does not seem to have occurred.
As ever, I welcome critiques and suggestions.