Here is my response to Steve Schaffner’s comments, which he kindly posted earlier based on a short extract from my first 2008 article.
The first contribution by @glipsnort is in a post that has had to remain in the previous thread, so I will quote the relevant part here in italics and interleave my responses:
@glipsnort: "Not exactly my area; I was an author of the chimpanzee genome paper, but my contribution was in modeling selection, not in the sequence comparison. On the other hand, I am pretty familiar with the paper, and it’s safe to say that the quoted text is completely wrong. Similar claims have been repeatedly introduced into Wikipedia, where they have have had to be weeded out.
I am very grateful to have a response to my old articles from you, Steve.
I was not aware that these figures had been repeatedly introduced to wikipedia and repeatedly weeded out. I think that the reason this happened was not because I had said anything from any position of authority, but because I (or perhaps others, if others spotted it) pointed something out that people could see from themselves in the figures given in the chimp genome paper. The paper clearly said how long the alignment was that the authors had made between the human and chimpanzee genomes. It was not at all obvious to the scientifically-literate reader what the reason was for the reason for non-aligning regions (apart perhaps from the reasons I pointed out in my 2008 articles). I am have to admit that I disappointed that references to this were apparently “weeded out” from Wikipedia rather than corrected with adequate and clear explanations of what was going on with the unaligned regions (please correct me if this was done - if it was I was not aware of it). If this had been done, this might have dealt with it more effectively.
@glipsnort: Short summary of the actual comparison:
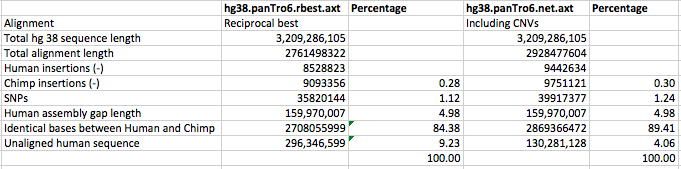
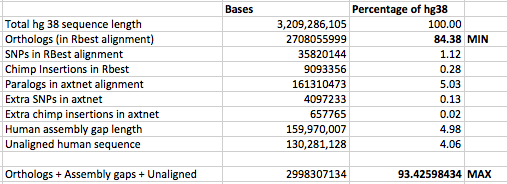
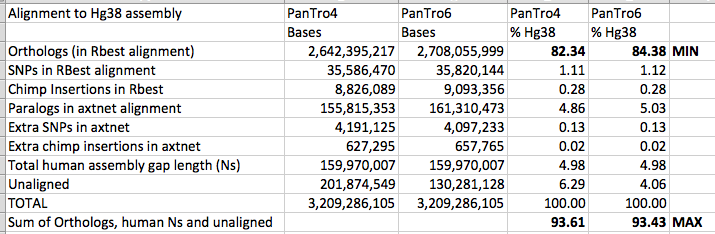
2700 million base pairs (out of a total of roughly 3100 million bp) of the chimpanzee genome was sequenced well enough to be compared. That is the portion we can say something about. Of that 2700 million, 2400 million could be aligned to the human genome. This portion is the basis for the conclusion that 1.23% of sites in shared DNA differ by a single-base substitution, and that another ~1.5% was unique to each genome.
I fully agree with these figures, and believe I cited them correctly in my articles.
@glipsnort: (Based on these numbers the most reasonable single statement of overall similarity is that approximately 97.3% of the human genome is identical to the chimpanzee genome.)
I think that is only a reasonable statement if it is accompanied by the caveat that this is based on only 2.4Gbp of the human genome. It is not good to cite a percentage without giving a sample size. Especially if not to do so is to imply that one’s sample size is the whole population when it is not. This is the heart of what this debate is about. I think one thing we can probably all agree on is that a single figure statement is not adequate alone, and needs to be accompanied by further explanation.
@glipsnort: The remaining 300 million base pairs of chimp DNA was sequenced but was not compared. 240 million bp were left out because they aligned to multiple places in the human genome. Much of this (I don’t know exactly how much) was the result of badly assembled chimp DNA, while some may represent genuine duplications in the human lineage. Another 90 million bp didn’t align to human at all; again, most of this was probably garbage of various kinds – badly assembled chimp DNA, parts of the human genome that hadn’t been assembled, etc."
This tells me what happened to parts of the chimp genome that did not align to the human genome, but does not tell me explicitly what is happening with the parts of the human genome that do not align to the chimp genome, apart from sequences that appeared to have copy number variation in human but not chimp. It is the parts of the human genome that do not align to the chimp genome that my articles were about.
In a later post @glipsnort says:
As I noted above, there was never 24% of the human genome that does not line up with the chimpanzee genome; most of that 24% represents DNA we never assembled in the chimp genome, so there was no comparison to be made.
The alignment of the human and chimpanzee genomes in 2005 only covered approx 76% of the human genome, so there was approx 24% of the human genome that did not align to the chimpanzee genome assembly. I am not quite sure what you are saying here, Steve. Are you saying that sequence that could align to the remaining 24% of the human genome is all actually in the real chimpanzee genome, and was sequenced but not assembled in 2005? If so, what evidence can you cite for this? If that is not what you are saying, please could you spell out your point more clearly?
@glipsnort: Who were the scientists who argued that this was junk DNA?
I did not find these unaligned regions being discussed anywhere in the literature (if they were, and I missed it, I would be very grateful if you could point me to relevant citations). This statement was based on conversation I had with scientists about the unaligned regions. It was not an entirely unreasonable suggestion as proliferation of a novel retro-element could generate a large amount of unalignable sequence relatively quickly and easily.
In conclusion, Steve, unless I am missing something, or misunderstanding something, you have not explained why, in 2005, I was “completely wrong”.
{kind=link}