Hi all,
I have done some more calculations for you to seek to come up with an upper bound of similarity between human and chimpanzee genomes, as well as a lower bound. This is based on the most recent alignments between human and chimpanzee genomes, available at the UCSC genomics website (hg38 versus PanTro6) see Index of /goldenpath/hg38/vsPanTro6
By my calculations, the human genome has between 84.4% and 93.4% one-to-one orthology with the chimpanzee genome. The uncertainty makes allowance for possible current incompleteness in our knowledge of both the human and chimpanzee genomes. The upper bound of 93.4% assumes that all the regions of the human and chimpanzee genomes that we have not yet assembled and/or aligned will prove to have one-to-one orthology between humans and chimps. The lower bound of 84.4% assumes that these regions will prove to be different between humans and chimpanzees. I have assumed throughout that further sequencing is unlikely to significantly alter currently known differences due to SNPs, indels, and copy number variation.
Here is how I did the calculation. I downloaded both the reciprocal best (“rbest”) alignment, and the “net” alignment (this allows copy number variation from UCSC for hg38 and PanTro6). Using custom PERL scripts, I measured the length of each alignment, and the number of SNPs and insertions in each one. I looked up the “Total assembly gap length” in the Hg38 human genome assembly statistics online (only a negligible number of these were present as Ns in the alignments). These are only known rather approximately, and many are estimated to the nearest 1000 or 10,000 bases.
Here are the stats I calculated for each alignment:
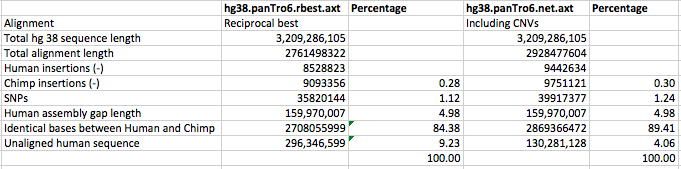
I used the differences between the two alignments to work out how much of the net.axt alignment was due to copy number variation, and how many SNPs and insertions there seem to be within copy number variants.
This yielded the following statistics for the overall similarity between the human and chimpanzee genome:
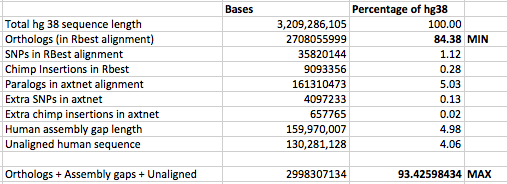
In my view, the upper bound of 93.4% is unlikely to prove to be the true value, once we have complete assemblies for both the human and chimpanzee genomes. This is because the regions of genomes that are hardest to assemble tend to be areas that are very repetitive, or fast evolving, or both. I therefore think it is unlikely that the 4.98% of the human genome that is represented by gaps in the hg38 assembly will prove to be orthologous to the chimpanzee genome. In addition, not all regions of the human genome that currently have no alignment look as if they are highly repetitive (though some are). I think that if these non-repetitive regions were present in the chimpanzee genome they would have been successfully sequenced and assembled by now.
I welcome feedback on these calculations and the methods and assumptions behind them, and especially identification of any errors I may have made.
Best wishes,
Richard