Thank you for that link – I hadn’t been aware of their response. Since most of it is in response to my comments, I’ll try to offer my own responses as I have time.
1 Like
Okay, let’s take the responses one at a time. My original comments are here.
Hössjer writes,
That is not an accurate summary of what I said. Here’s what I actually wrote: “For example, it cites ref. 49 to claim that Y chromosome diversity is smaller than previously believed. Ref. 49 doesn’t say that at all, and in reality the relevant Y chromosome diversity – the length of the oldest branches – was shown in 2013 to be much higher than previously thought, when a new, highly divergent branch was discovered.” The referenced study is about why diversity is low on the Y chromosome, not about the Y’s diversity being lower than previously believed. As I noted, estimates of diversity on the Y have in fact increased in recent years, not decreased.
No, I didn’t offer a critique of Sanford’s paper, since it was incidental to the subject at hand. Sanford models the time for a string of specific mutations to occur, i.e. it assumes there is an extremely specific target for evolution. That process almost never corresponds to real evolutionary processes, which have no target, which can go in a vast number of directions, and which typically have numerous route to achieve a specific phenotype. Those are the processes that evolutionary biology seeks to model. For example, just in humans, multiple routes, involving independent mutations, have been taken to achieve short stature, lactase persistence, reduced pigmentation, increased pigmentation, altered fat metabolism, malaria resistance and high altitude adaptation. Lighter skin color alone involved at least several independent mutations in multiple lineages. If Sanford’s model were an accurate representation of the adaptive process, it should have taken tens of millions to billions of years for lighter skin to emerge. In reality, it took a few thousand.
The response doesn’t really address the problem. The frequency spectrum of human genetic variation is very close to falling off as 1/f, with lots and lots of moderately rare (say, 0.1% to 20% frequency) variants seen. The frequency spectrum of genetic variation inherited from a first couple would peak at 25% or higher. The observed spectrum looks like very much like what you would expect from a more or less constant sized population accumulating mutations for a very long time. More importantly, it shows no evidence at all for a contribution from an initial couple.
Now, it is certainly true that the further back in time you postulate the initial couple, and the smaller you make their contribution to modern genetic variation, the smaller the discrepancy becomes between the model and the observed variation. Eventually you will remove any discrepancy, when you get to the point that the initial couple contribute essentially nothing to observed diversity. So sure, you can match the observation with the authors’ model – provided you turn their model into a more or less constant sized population that’s been accumulating mutations for a very long time, and to which Adam and Eve contribute little or nothing. But in that case, what’s the point of the model?
There are lots of factors that influence the allele frequency spectrum. No population history, though, will turn genetic contributions from Adam and Eve into a 1/f spectrum. In mathematical terms, population history cannot change the expectation value of the frequency of an allele.
Reference 24 doesn’t help. It’s talking about rapid population expansion producing rare alleles from new mutations. For the variation contributed by Adam and Eve, which is the focus here, rapid expansion will simply freeze the initial frequencies into place, increasing the discrepancy with the observed distribution.
I don’t have time to go into great detail, but here are some relevant data. Take a look at the 22 human autosomes (all chromosomes except for the sex chromosomes). If human diversity is the result of accumulated mutations, we would expect regions with a higher mutation rate to have more diversity. Assuming mutation rates don’t change very quickly, we would also expect to see larger genetic divergence between humans and chimpanzees in those regions, since that divergence also represents accumulated mutation. Therefore, we should see a correlation between human diversity and human/chimpanzee divergence. We do. Here I’ve broken the genome into 1 million-base windows, and plot the human/chimp divergence for that window on the x axis and human diversity in that window on the y axis:
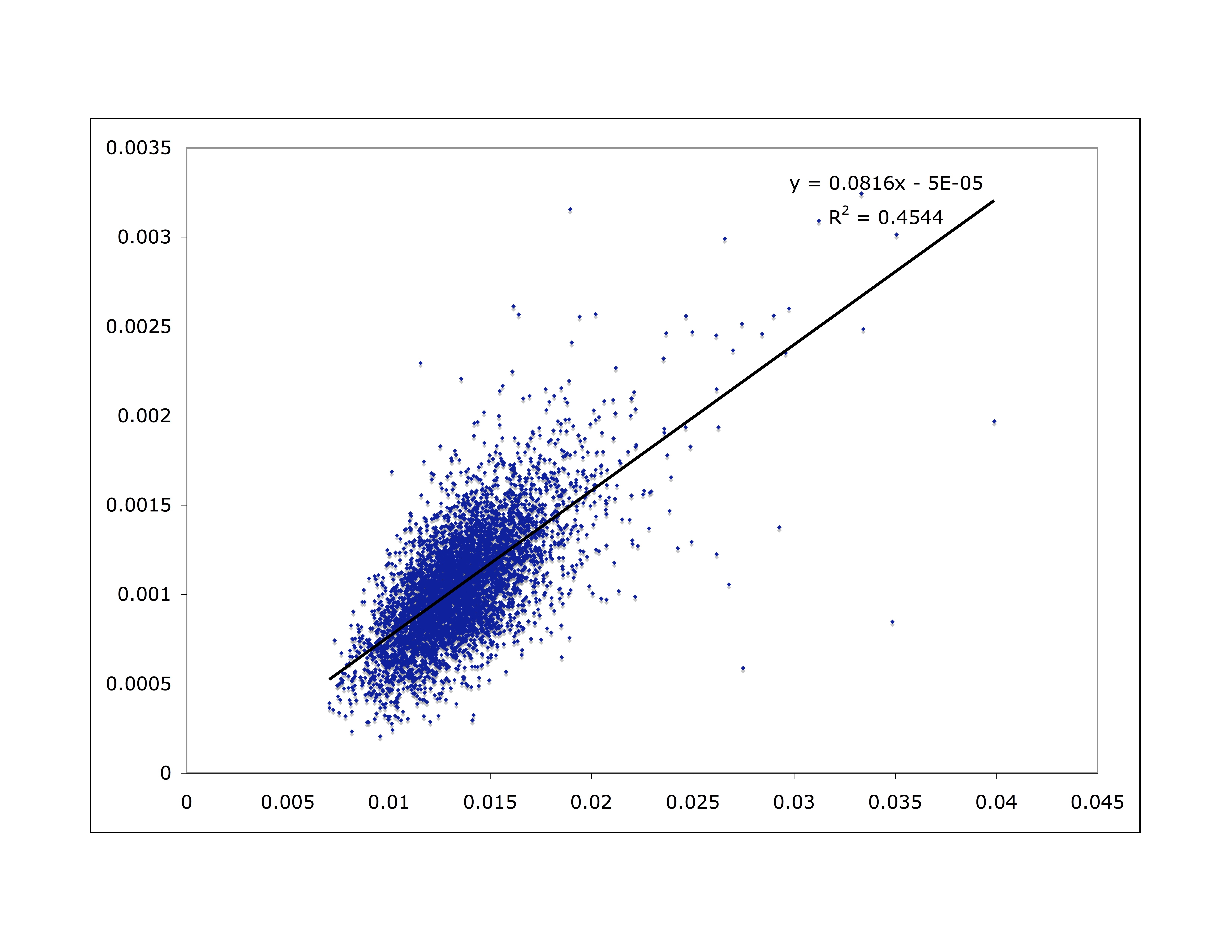
As is readily apparent, there is indeed a correlation.
You can also look at the kinds of mutation. Some mutations happen rarely (e.g. C->G), while others happen more often (e.g. C->T, because C and T are chemically similar), while others happen very often (C->T mutations when the C is followed by a G on the chromosome, for well-understood chemical reasons). If genetic diversity results from accumulated mutations, when we look at genetic variants we should see more of the common mutations and less of the infrequent ones. That is indeed what we see. We should also see the same pattern repeated in human/chimpanzee genetic differences. Here are the two distributions:

The top graph shows the distribution for human/chimpanzee genetic differences, and the bottom shows human/human differences. It would be hard to argue that these patterns, and their similarity, are just a coincidence.
[quote]
However, Ann Gauger has pointed out that chimps have very different block boundaries than humans as another argument against common descent.[/quote]
I would like to see that argument fleshed out. Human block boundaries are largely determined by recombination hotspots, whose locations are largely determined by a particular DNA motif – a pattern of DNA bases that a particular protein likes to bind to. A small change to that protein can cause it to bind to a different motif, thus completely changing the pattern of hotspots across the genome. Now we have good evidence that that protein (which is coded for by a gene named PRDM9) changes frequently, mostly because all the recombination at hotspots eventually destroys the hotspots, and it becomes selectively advantageous to switch to a new motif. Thus it is not at all surprising that chimpanzees should have a different PRDM9, and thus different hotspots – especially since we see mutated forms of PRDM9, and different hotspot locations, even within humans, with the mutated forms apparently being under positive selection.
In sum, I don’t believe Hössjer has successfully addressed any of my criticisms of their paper.
5 Likes
This topic was automatically closed 6 days after the last reply. New replies are no longer allowed.