Okay, here are my current thoughts on trans-species variation. I invite a deep dive in the literature to see if anyone can find a key paper I overlooked. Please prove me wrong if you can…
Trans-species variation. The evidence against an ancient bottleneck in trans-species variation is not as strong as I had thought.
As we have seen, there is a limit how far back the evidence from Human Variation gives us confidence against a single couple bottleneck. Before about 500 kya, it is possible that such a bottleneck, if brief, would be undetected in by current population genetics models. The specific number may be adjusted upwards by further analysis, but it’s a good starting point for now.
However, this is not actually the strongest argument put forward against a single couple bottleneck since we diverged from chimpanzees. For that, we have to look more closely at Trans-Species Variation.
Trans-Species Variation
Human Variation and Trans-Species Variation are related but different. To measure human variation, we look at a large number of human sequences. To measure trans-species variation, we look at a large number of both human and non-human sequences, usually chimpanzee. From looking at this data, we might find evidence of alleles that appears both in chimpanzees (for example) and humans.
This figure illustrates what appears to be happening:
Trans-species polymorphism in humans and the great apes is generally maintained by balancing selection that modulates the host immune response | Human Genomics | Springer Nature Link
The key point is that along each of the colored lines, several lineages are being shared between different species at a single place in the genome. Normally, there would be just one lineage on these time scales, but balancing selection maintains multiple lineages of alleles. By counting the number of allele lineages shared between humans and others, we can put a hard-stop lower bound on a bottleneck going back before humans and chimps diverge. Whatever bottlenecks there are they have to be big enough to include all the trans-species lineages.
Molecular Clock Not Valid
One tempting argument, which is not quite right, is to just estimate the TMRCA (or TMR4A) of these alleles, the same as we did across the genome, and use this as an estimate of a bottleneck time. This however, is an error.
Something called “balancing selection” is critical for enabling variation to last long enough to be shared this long between humans and other species, and this usually happens in proteins important for our immune response. So we see trans-species in only a few regions of the genome.
However, balancing selection violates the conditions required to accurately date variation in DNA. We cannot use our formula D = R * T here, because, in this case, we do not have a valid way of estimating R over these time frames. While in neutral regions of the genome, the average mutation rate works in our favor, at times we expect balancing selection to be increasing the rate of change in unpredictable and untestable ways. This can happen very rapidly as balancing selection can even select for increased mutation rates within this region.
Ayala’s Argument Against a Bottleneck
The argument here is two part. First, from effective population size estimates, and second from trans-species variation. I’m not going to engage the argument about effective population size, because it appears to be incorrect. Very tight bottleneck can still have high effective population size, and it seems Ayala missed this point. But this just takes us back to the TMR4A work.
This is where trans-species variation becomes important. It gives an independent way of dating alleles. If an allele in humans is closer to non-human alleles, it appears that it existed before those two species diverged, and was maintained by balancing selection to this day.
This study by Francisco Ayala was the first, to my knowledge, to make the case against a bottleneck by studying trans species variation HLA alleles. The Evolution of Human Populations: A Molecular Perspective - ScienceDirect
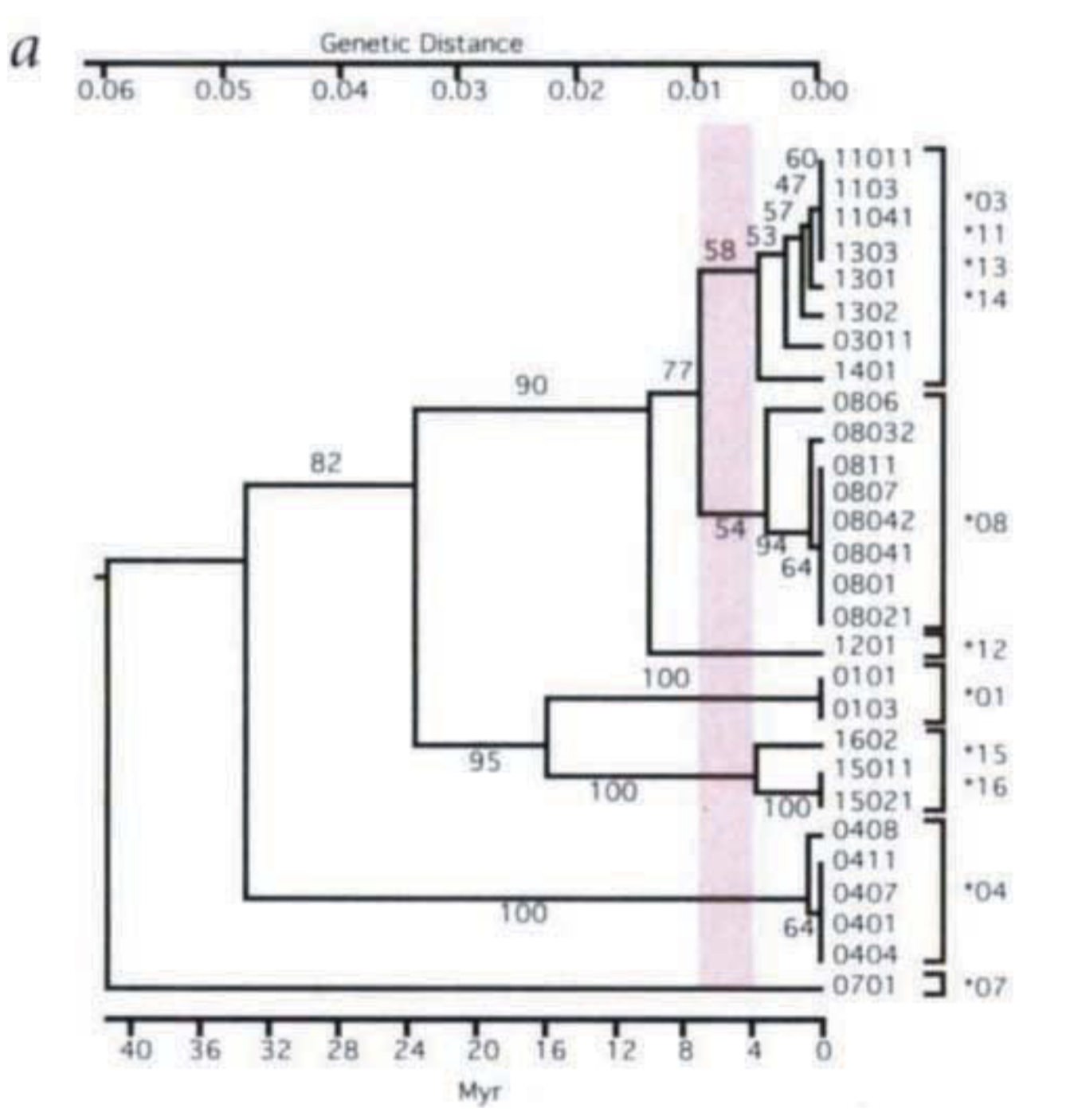
This figure from Ayala shows human alleles with other primate alleles joined by similarity, not phylogenetic analysis that respects nested clades. I’ve highlighted the human alleles in this figure, and drawn red circles around 7 clusters of alleles which appear to be shared between human and other species. Remember, we can only put 4 alleles at each position in the genome of a couple, so this seems (at least on face value) to demonstrate there must have been at least 4 individuals in the tightest bottleneck of our ancestors.
Ayala’s summary is:
Figure 4 is a genealogy of the HLA alleles obtained by the UPGMA method, which assumes constant rates of evolution and thus aligns all 19 alleles at the zero- distance point that corresponds to the present. The ge- nealogy suggests that 8 allele lineages were already in existence 15 Myr ago, at the time of the divergence of the orangutan from the lineage of African apes and hu- mans; and that 12 allele lineages were in existence 6 Myr ago, at the time of divergence of humans, chimps, and gorillas.
The difference between his numbers and mine in how we determine lineages. There is some ambiguity in how we determine the cutoffs. Still, as long as we see more than 4 lineages with trans-species variation, its seems like evidence against a single couple bottleneck. From this, he argues,
There is, however, no evidence supporting the claim that ex- treme bottlenecks of just a few individuals, such as postulated by some speciation models (Mayr, 1963; Car- son, 1968, 1986), have occurred in association with hominid speciation events, or with major morphological changes, at any time over that last several million years.
This is probably correct, in that there is no evidence for a bottleneck that I can see. But he means here to mean that a bottleneck has not happened: i.e. there is evidence against a bottleneck in the last several million years. That may be incorrect.
Some Technical Asterix
Generally speaking, this work has been understood in the field to definitely discount any notion of a single couple bottleneck. On face value, that is certainly what it looks like. However, there are some big caveats.
- The molecular clock based dates computed in these studies, it does not appear to be well calibrated.
- We do not really know the confidence on any of these clusters, because Ayala did not estimate them using modern bayesian methods.
- He also used a similarity based method to build the trees, rather than a true phylogenetic reconstruction. This is important, because it can produce different clusters.
- It does not appear convergent evolution was accounted for in this analysis. Convergent evolution, at this level, can create the appearance of shared history when there is none.
- His population simulation used a bottleneck lasting 10 generations (e.g. 10 individuals for 10 generations), which is much longer than the bottlenecks we are considering (e.g. 2, to 10, to 500, to 2500, to 12500).
While these are interesting results, at some point, this analysis needs to be done with better methods to really determine how many lineages are persistent over the last 6 mya. Moreover, effort to correct for convergent evolution is important here too. On the simulation size, a brief bottleneck needs to be considered, rather than just those of 10 generations.
A Finding Not Replicated
Ayala focused his work on HLA-DBQ1 (one of the MHC genes), but similar work has shown trans-species variation at other locations in the genome. However, I could not uncover a single other study that shows more than 4 lineages with tran-species variation.
I cannot do a full review here, but we can see the balancing at other genes, with fewer lineages in the end. For example…
Common chimpanzees have greater diversity than humans at two of the three highly polymorphic MHC class I genes - PubMed
This figure is fairly typical of findings…
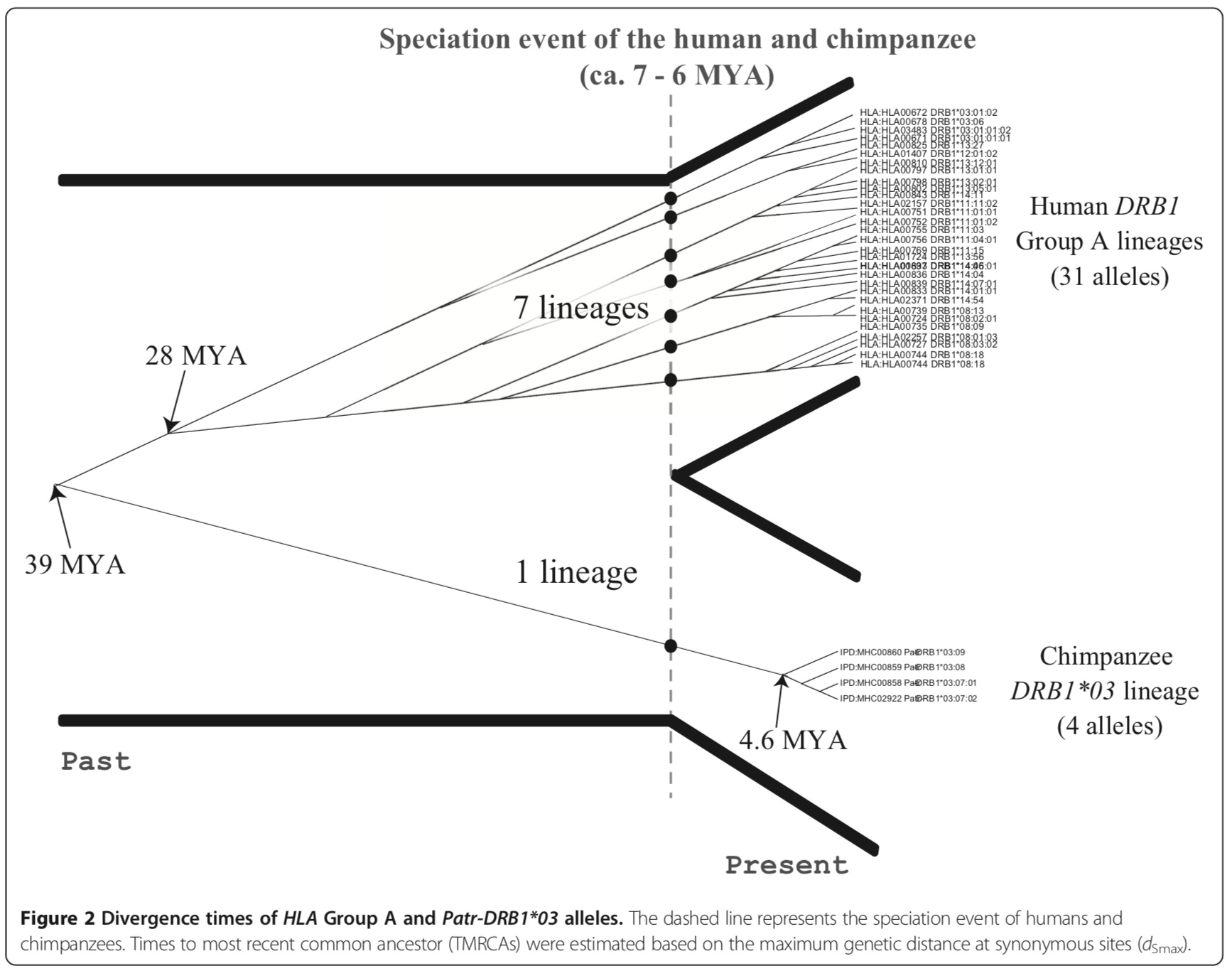
A human-specific allelic group of the MHC DRB1 gene in primates - PMC
This figure shows a molecular clock based estimate (which do not appear well-callibrated) of 7 lineages at 6 mya, however, less than four lineages (0 in this case) is shared with chimpanzee. Reviewing several papers, I cannot find replication of Ayala’s findings of more than 4 lineages being shared between humans and other species.
We can see this pattern in this figure too…
A human-specific allelic group of the MHC DRB1 gene in primates - PMC
Here, the bold leaves are human sequences. Notice the difference between this figure and Ayala’s. There is numbers on the edges (which indicate confidence) and we just do not see nearly as many lineages in common. The authors here conclude there is just one lineage in common.
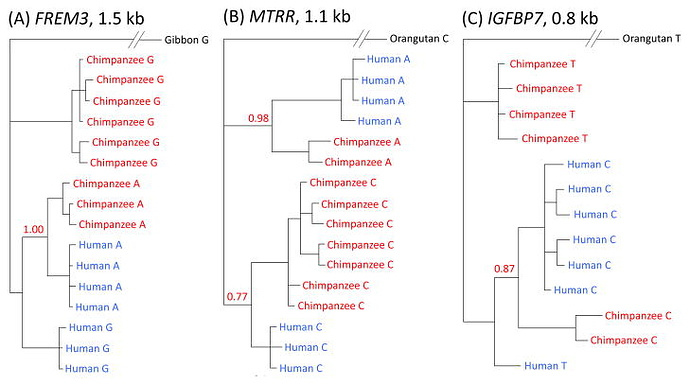
Here is another typical results figure:
Multiple instances of ancient balancing selection shared between humans and chimpanzees - PMC
Each tree is a different region of the genome. Notice, again, that there does not appear to be more than 4 clusters with both human + chimpanzee alleles.
While Ayala is an established scientist, his work was done in 1996, well before modern sequencing efforts, and modern bayesian analysis of phylogenetic trees. While no one has published on DBQ1 since he did, it is very surprising that no one else has replicated his result in the last 22 years on another locus. Of course, if someone can find a study that does, please let me know!,
The apparent failure to replicate this finding (with (1) much more data, and (2) improved methods), discounts substantially my trust in his findings. We just know much more about how analyze these sequences, and we have so many more of them. It is not surprising that our understanding might advance.
One Line of Evidence? One Paper?
At the moment, the Ayala paper appears to be the only study which shows more than 4 allele lineages with trans-species variation. His analysis, however, did not estimate confidence nor did it use phylogenetics to determine lineages. In 22 years, I cannot find a paper that replicates his finding. Certainly, trans-species variation has been observed, but not more than 4 lineages, as far as I can tell.
This is not enough evidence by which to make a confident claim against a single generation bottleneck.
The Way Forward
The right way forward, then, is to to study trans-species variation with the data we have now, but better methods than did Ayala. This takes some difficult work, however. I’m not 100% sure if we will give it a try here, but we might. This, also, is the most likely place a future study might uncover evidence against a single couple bottleneck.
Until that happens, however, I am not sure this is strong evidence against a brief bottleneck. I stand to be corrected, however, if someone can produce a study that shows this. If you find one, please send it to me.