The empirical data I’m trying to match is purely from current population data. The mutation rate is a fixed parameter in the model. It can be estimated from comparison with ancient DNA, or from comparison with another species, but the estimate I’m using is based on data from modern populations. The generation time also comes from data on modern populations.
Thanks for this
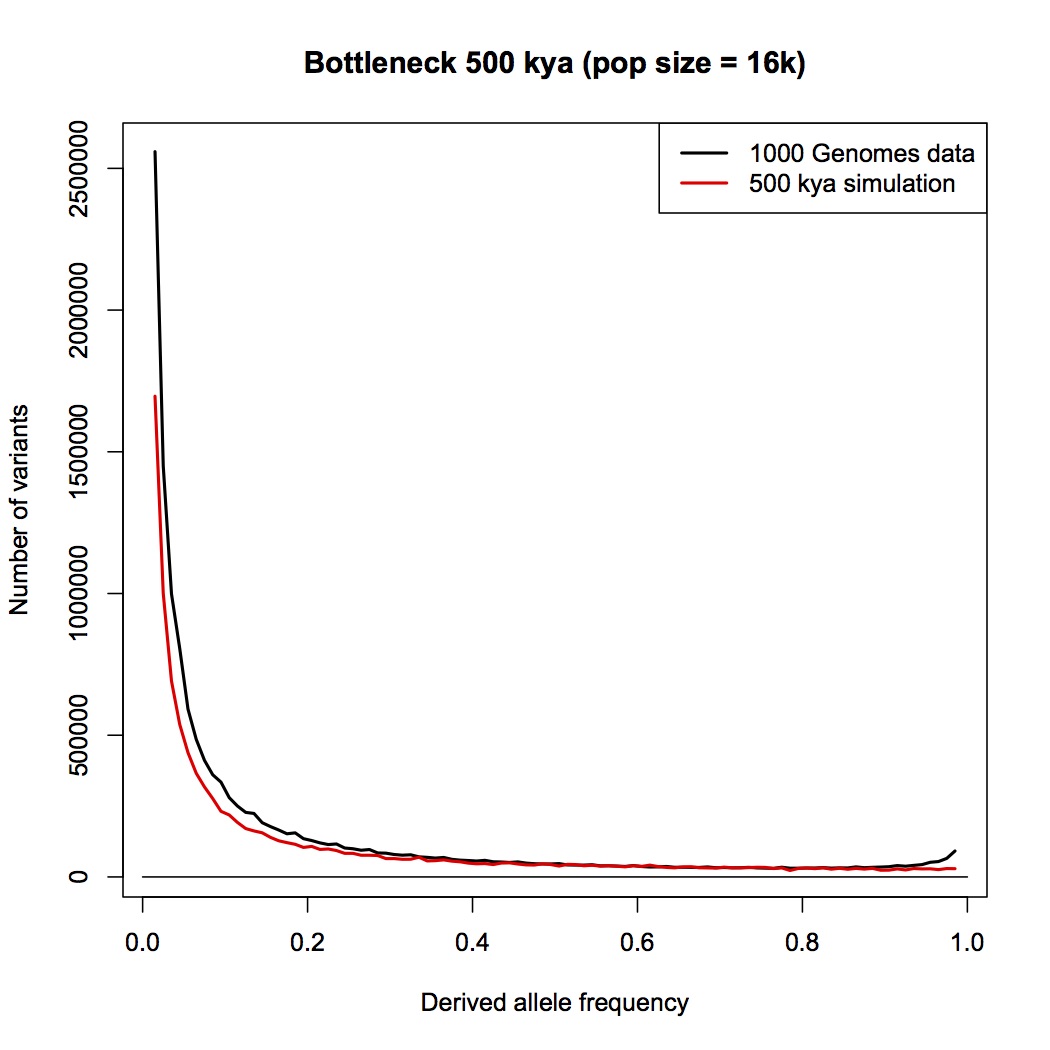
Should the legend say 500 kya simulation instead of 100?
Classic question: how large would the error bars on that plot be, given the uncertainty in the mutation rate estimate? Don’t bother if it costs effort to figure out the answer, I’m already amazed that you have time on your hands to run these simulations.
2 Likes
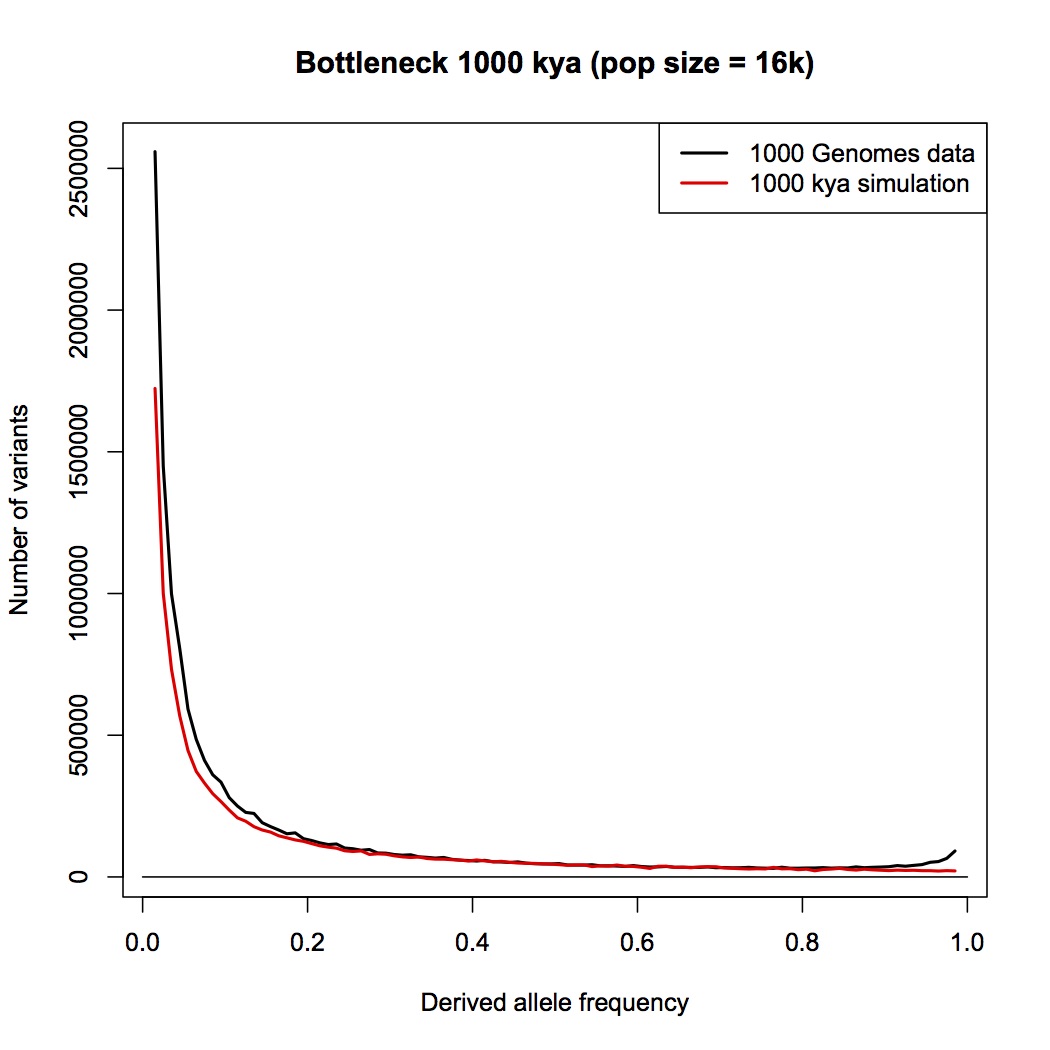
1 million years. By eye, this doesn’t look any different than the constant-sized population compared to data that I posted earlier. Add a population expansion and it would fit well.
2 Likes
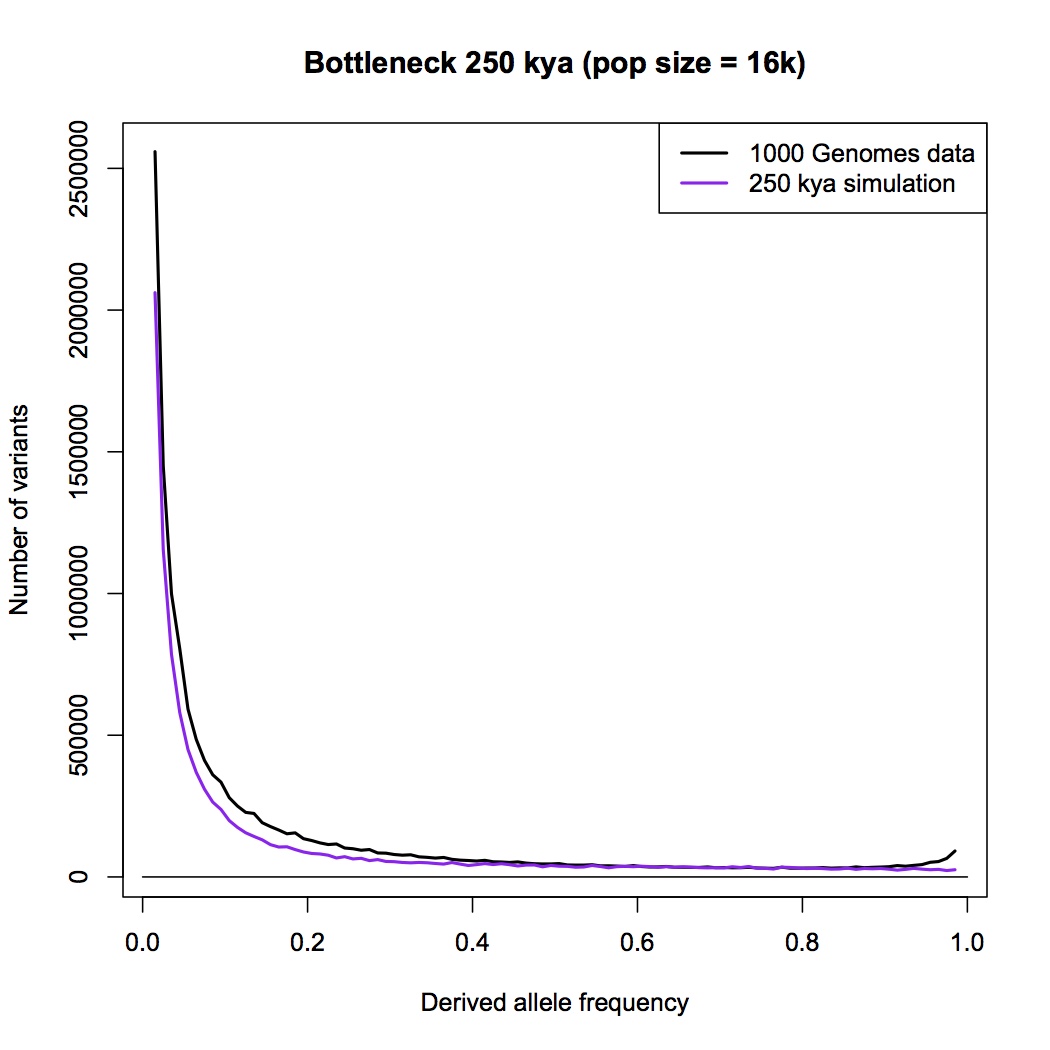
Not a simple question, in this case. The mutation rate is poorly known, and why it’s poorly known is also not well understood. Estimates range over roughly 1.1 to 2.0 x 10^-8; most recent estimates have been near the low end of that range. To a varying extent, changing the mutation rate can be absorbed by changing the population size – completely for a constant-sized population, less so for genetic drift after the bottleneck.
What I have done is run a couple of the simulations using the highest mutation rate within the above range (2.0 x 10^-8). Here is 250,000 years, pop size = 16,000. You can compare it to the plot above for the same age, which was done with mut rate = 1.6 x 10^-8.
1 Like
Thank you so much for doing these analyses, Steve. I was hoping that my Nature Eco Evo blog would stimulate some studies that set out to explicitly test the bottleneck of two hypothesis, and this is certainly a big step in that direction.
As I begin to comment on this, I think I should say for those reading in who are not in the science world that Steve Schaffner is right at the top of the field when it comes to human genomics, and was one of the authors of the 1,000 genomes paper (and many other highly cited and very significant papers too). It is a real privilege to us all who are interested in this issue to have Steve running simulations on the two person bottleneck hypothesis, and to be taking the time to answer questions on it.
I would also note that the fact that we are discussing these new simulations is in itself very good backing for the point I made in my blog that more research is needed on this issue. It highlights how mistaken it is to declare that we can be as certain that there has not been a two person bottleneck as we can be that the earth rotates around the sun. After all, if I were to question the latter, no one would need to go away and do a simulation to come up with new evidence for it, in order to be persuasive.
Steve, I am very interested in your analyses. I had expected allele counts at polymorphic loci to be the biggest argument I would come across against the bottleneck of two hypothesis. I was not expecting an argument from allele frequency spectra. I am delighted to come across this possible way to test the hypothesis that I had not thought of, and that was not mentioned in Dennis’ book chapter.
I am still going to take a bit of convincing that this is a good approach to testing the hypothesis, however. I will explain my reasoning below. I would underline that I know you see what you have done as just a preliminary study and you yourself are well aware of the approximations and simplifications that you have had to make. I will try to explain my points as simply as I can for our readers.
-
Steve has already highlighted that this approach depends heavily on a correct estimation of mutation rates, and the model presented assumes that these do not vary with time or in different parts of the genome. This may not be the case in reality.
-
Also, as far as I can see (Steve, do correct me if I am wrong), this approach depends on the assumption of a single panmictic population over the timespan that is being examined. I think it would be fair to say that there has been substantial population substructure in Africa over that timespan and that this has varied over time. To my mind, this population substructure could well boost the number of alleles at the frequencies of 0.05 to 0.2.
Let me just try to explain that in a way that is a bit more accessible to our readers. I am saying that Steve’s model (at least in its current preliminary form) is making the approximation that there is one single interbreeding population that has been present in Africa throughout history, and that mating is random within that population. However, the actual history is almost certainly very different to this. The population would have been divided into smaller tribal groups which mainly bred within themselves. Within these small populations, some new mutations would have spread to all individuals and reached an allele frequency of 100%. In other tribes these mutations would not have happened at all. Thus if you treated them all as a large population, you would see an allele frequency spectrum that would depend on how many individuals you sampled from each tribe. It is more complicated than this because every-so-often tribes would meet each other after a long time of separation and interbreed, or one tribe would take over another tribe and subsume it within itself. Such a complex history, over tens or hundreds of thousands of years would be impossible to reconstruct accurately, but would distort the allele frequency spectrum away from what we would expect from a single population with random mating. It gets even more complicated if we start also including monogamy, or polygamy.
-
As far as I can see the model currently also assumes no admixture from outside of Africa. A group of people arriving in Africa from another continent would affect the allele frequency spectrum if they interbred, and if their non-African population had diverged from African populations. Obviously this could not have happened at time periods when there were no humans outside Africa. But the data under analysis is obviously of present day Africans after centuries of admixture from outside Africa. Steve may be able to account for this with a more complex model that excluded alleles that are common in non-African populations, although it would be hard to be completely sure about the origins of these alleles.
-
As far as I can see, the model currently assumes no selection. Natural selection will boost the frequency of beneficial alleles (and alleles linked to an allele being selected for). Especially relevant would be alleles selected in one location and not another, and alleles under balancing selection. Steve would know better than me how to try to incorporate selection into the model, but my guess is that it would be very tricky.
Finally, could I ask, Steve, how many allelic variants did you assume in the founding couple, and what proportions of alleles did you put in them at 25% and 50%? Or did you assume that all variants arose through mutation?
2 Likes
@glipsnort. Thanks very much for doing this. When I asked about it being computationally intensive I had forgotten you only let the population double up to 16K and then held the population fixed.
We have planned to test the effects of varying various parameters, as you suggest. No more requests.
1 Like
@glipsnort @RichardBuggs @T_aquaticus
Richard, you have mentioned highly polymorphic genes as posing a major problem to the 2 person bottleneck hypothesis. Perhaps you and Steve are aware of this paper:
They surveyed the HLA-A, B, C and DRB1 and DQB1 loci from 6 million individuals, and found purifying selection at the level of haplotype for Class I genes but not Class II. There are clear pairings of alleles which co-occur; in addition recombination appears to be suppressed in the region. Then they looked for selection between alleles and found that new alleles were favored. It is at the level of the allele (usually exon 2) where mutation rates and gene conversion rates are thought to be high.
I’d be interested to get your opinion about this paper and its possible relevance to the bottleneck question.
I see the conversation has continued in my absence - I’ve been away from email for two days. Hunting, actually. Hauling a rifle up and down steep cliffs seeking elusive deer is remarkably therapeutic and very good exercise…
Steve, thanks for those simulations. Very nice.
Richard - I see you’re still at me for specific references. The paper that specifically undergirds the “pick a few genes” part of the broad-brush summary statement about allele methods is this one, which like PSMC, is a coalescent-based approach:
Darrel Falk and I also discussed that one way back when I first started writing for BioLogos.
Now, is PSMC an allele-based method? Does it “count” alleles? I guess it’s a bit semantic at this point - but that’s one of the challenges of writing for a non-specialist audience. Explain coalescence, or talk about it in simpler terms? I went with simpler terms. Sorry if it was confusing.
2 Likes
Also, for this paper, note that it gives an Ne at about 18,000, not the usual “10,000” - and it’s based on polymorphic Alu insertions, and thus not on the standard forward mutation frequency (Mu). So, it’s another independent measure of ancestral population sizes that again, does not (at all) support a bottleneck to two.
1 Like
Also note this from the paper: they considered, tested, and rejected a strong bottleneck hypothesis in the time frame we are discussing. Note how they cite the allele frequency spectrum for the Alu variant sites:
“The disagreement between the two figures suggests a mild hourglass constriction of human
effective size during the last interglacial since 6000 is very different from 18,000. On the other hand our results also deny the hypothesis that there was a severe hourglass contraction in the number of our ancestors in the late middle and upper Pleistocene. If humans were descended from some small group of survivors of a catastrophic loss of population, then the distribution of ascertained Alu polymorphisms would show a pre- ponderance of high frequency insertions (unpublished simulation results). Instead the suggestion is that our ancestors were not part of a world networkof gene flow among archaic human populations but were instead effectively a separate species with effective size of 10,000-20,000 throughout the Pleistocene.”
3 Likes
Really? Why don’t we conclude that the speed of light gets faster and faster with time while we are at it.
6000 years ago, it took 4 hours for you to see the stampeding wildebeasts… and naturally, you were run over by them before you could literally see them.
For you to assume that mutation rates were different, you would need evidence for why that would be, yes?
Conversely, if we correlated mutation rates across several types of animals and types of phenotypes… if we saw convergence and general agreement, your theory would be proved wrong.
So… instead of “what-if”-ing scientists to death … maybe you could collect the evidence that shows you something?
George, the quote you are making is from Dennis’ book, which I was quoting. I did not pen those words. However, I would point out there is a considerable literature on the evolution of mutation rate. Michael Lynch has done a lot of work on this. This is very different from the speed of light.
Hi Dennis, good to hear you have had a nice hunting trip. I trust you are enjoying some venison now.
I am indeed. I cannot underline enough how important this issue is. If you are making unsubstantiated or mistaken claims about science in your book, just lines after saying “given the importance of this question for many Christians”, I don’t think it is just me who views that as quite a serious issue. This is why I am so keen to give you every opportunity to substantiate this passage.
For readers struggling to follow all the different threads in this comments stream, let me remind them that this is the passage from Adam and the Genome that we are discussing
So far, these are the attempts you have made to substantiate this passage from your book.[quote=“DennisVenema, post:13, topic:37039”]
Some of the citations you’re looking for are just working familiarity with published data sets.
[/quote]
I have argued that the plain meaning of that passage in your book, backed up by your Part I blog, is that it is not a summary statement and not a reference to PSMC and that to merely refer to datasets without the described analyses is not an adequate citation.
I don’t know how much time you have had to read through all the posts since your hunting trip. To make sure that others would agree with me about the plain meaning of the passage from your book about allele counting methods, I posed three simple questions for others to answer about it.[quote=“RichardBuggs, post:35, topic:37039”]
The questions I ask you are, when you read the extract from Adam and the Genome in bold below, which I show in its context:
Does the passage make you think that it is referring to a scientific study where a few genes have been selected and the number of alleles of those genes in current day human populations have been measured?
Does the passage make you think that someone has done calculations on these genes on a computer that have indicated that the ancestral population size for humans is around 10,000?
Does the passage make you think that this is a different method to the PSMC method?
[/quote]
To which your Biologos colleague Ted Davis answered:
And another reader also agrees with my reading of the passage: [quote=“tallen_1, post:38, topic:37039”]
As far as I can tell, Dennis makes three claims most relevant to your point: One, that there is a method to estimate minimum ancestral population sizes based upon measurements of number of alleles across various genes present in a population, and that this method indicates a population of approx. 10,000. Two, that an independent method exists that does not rely upon estimates of past mutation rates, involving “linkage disequilibrium,” that converges upon the same ancestral population size of 10,000. Three, that there has been a more recent method that is similar (not identical) that is not independent of mutation rate but also converges on similar results, namely the PSMC method.
Of these three approaches, Dennis’s support for the first seems to derive mostly from calculations on collected data. Presumably done by himself or others. Of the latter two approaches, that does seem to be something that is published and to which he could (and I think did) direct you. But I’m unclear as to whether the published studies for the latter two methods explicitly state Dennis’s conclusions or if he is drawing as well primarily on their collected data for support. I’m perhaps at a bit of a handicap on this as I’m relying on only excerpts of his book here on this thread. But to your point I do believe he describes three distinct methods. I’m eager to hear more about the sort of calculations conducted in these methods and how they may or may not support Dennis’s argument. That is what I am looking forward to in his remaining parts to this topic.
[/quote]
No one, so far, has defended your reading of the passage. This is making me think that your reading of the passage is what you wish you had written, rather than what you actually wrote.
And now in your latest posts you are saying:
My first response was to think “Well, thank you, why didn’t you say so before?” But a quick skim of the paper convinces me that, again, this is not an adequate citation to support the passage we are discussing.
- It was published before the Human Genome Project, and before we had “sequenced the DNA of thousands of humans”
- Most of the genes (if we may loosely call a retrotransposon a “gene)” in the paper are monomorphic in the human population studied and a handful are dimorphic. Thus the maximum number of alleles at any locus in the study is two. The allele counting method as described in your book, and elaborated upon in your Part I blog, explicitly requires higher numbers of alleles.
So again, I don’t think this is an adequate citation.
Dennis, I have to say the conclusion I am coming to is that you made a mistake in your book. If so, I would have huge respect for you if you were willing to admit it, then we could all move on and discuss the interesting science of the other methods you have written about, and the work that Steven Schaffner is doing. We all make mistakes, and those of us active in research are very used to having them forcibly pointed out to us when we get back peer review comments on our manuscripts and grant proposals. It is never much fun to have them pointed out, but part of being a good scientist is being willing to correct our mistakes and move on.
1 Like
Richard, I agree the passage is not clear. My mistake was trying to shove too much into a short summary in a way that would be accessible. I was over my word count as it was and things needed to be concise. Obviously that part was too concise to the point of confusion.
Like I said, it’s my (in hindsight poor) attempt of a summary of the field as a whole, for all allele-based methods.
I’m not sure why you continue to insist that that summary excludes PSMC methods. It doesn’t. I was primarily thinking of the 1000 genomes papers, but also all of the older literature prior to the human genome project work. Are you really saying that you know better than I what was in my mind as I wrote that passage?
What should be a bigger issue than my unclear writing is that there is no evidence in the literature that supports your hypothesis, and plenty of evidence that supports my conclusion in the book - which is the whole point that that passage is trying to convey, albeit in too compressed a fashion. Early work and the massive results from the Human Genome Project agree: humans are too diverse to have come from just two people.
So yes, by all means, let’s discuss the science. How about that Alu paper? It specifically tests the hypothesis you’re asking about, which counters your claim that researchers have not considered your hypothesis. Do you think that Alu polymorphisms in present-day humans are compatible with a population bottleneck to two people within the last 200,000 or 300,000 years? Why or why not? My take on it is a resounding “no”. It’s also nice that it’s not based on the nucleotide mutation rate, so it provides a check against papers that have to estimate that. It fits right in with the allele frequency spectrum data for SNPs that Steve is laying out for you. If there had been a human bottleneck, we would see skewed frequency spectrum for Alu insertions as well as for SNPs.
After we’re done discussing that paper, we can also discuss this other older one if you like (and then the 1000 genomes papers, and so on):
And since we’re on the topic of the pre-genome project literature, here are two other papers that I consider “older” papers, although they are sort of genome project papers, since they were published based on studies on specific genome regions when the HGP was underway/nearly done. These papers also were part of the older literature that formed my understanding of the data, and they are based on allelic variation in small genome regions. Even though they are older, they remain relevant. One even explicitly says there was no severe bottleneck in the last 500,000 years.
And last, but not least, another early paper that is part of the body of knowledge of the field as a whole.
https://academic.oup.com/mbe/article/10/1/2/1030040
These last three papers are also under the surface of the “pick a few genes” statement, FYI.
Edit: a few more early papers, also part of the discussion. Remember, that summary statement is a gloss of the field using allele diversity methods. There might be other papers I’m not remembering at the moment too, but these at least give a sampling.
1 Like
Dennis,
As a non-specialist here, I’m doing my best to follow along. But I’m hoping you can clear things up.
In my response to Richard above, I noted you seem to provide three distinct methods to estimate a minimal bottleneck size in human population over the past (presumably) hundreds of thousands of years. With one of them being independent of mutation rate. And all three converging on the same result. In the back-and-forth on this thread, I’m having a difficult time separating evidence and claims presented into these three methods. It seems to muddle together a bit. Again, as a non-specialist. I’m also struggling to gain clarity on how some of the studies tossed around here connects to the distinct methods you discussed in your book, and whether they explicitly back up your claims for at least the latter two methods or if there is some chain of inference and extrapolation that distances us somewhat from the authors’ conclusions in those studies. And perhaps better understand exactly how you intended all of that to come out. Again as a non-specialist. So I’m doing my absolute best here, but am hoping for some help. I presume most other readers with passing familiarity with the material, but no professional expertise, may find themselves in a similar quandary. Looking forward to your thoughts. Thanks Dennis!
1 Like