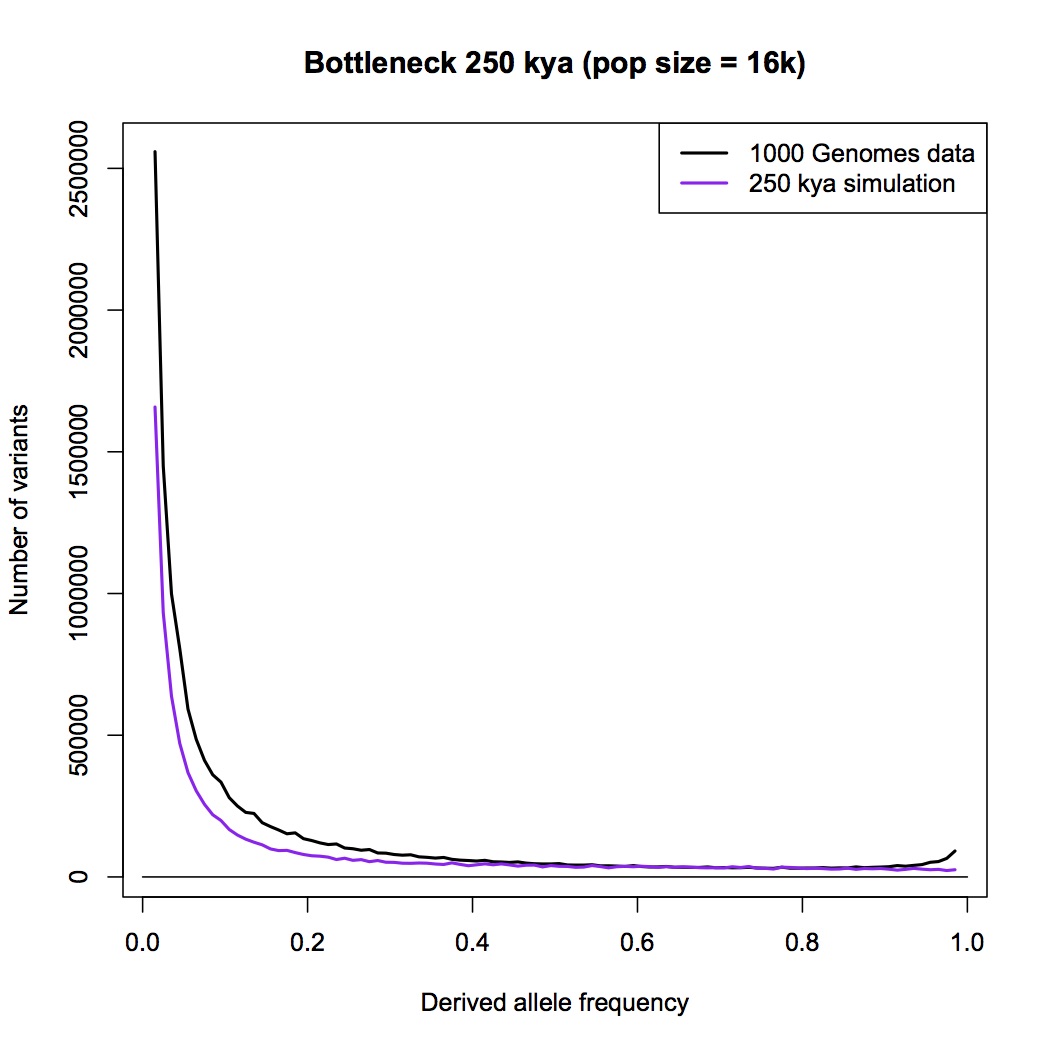
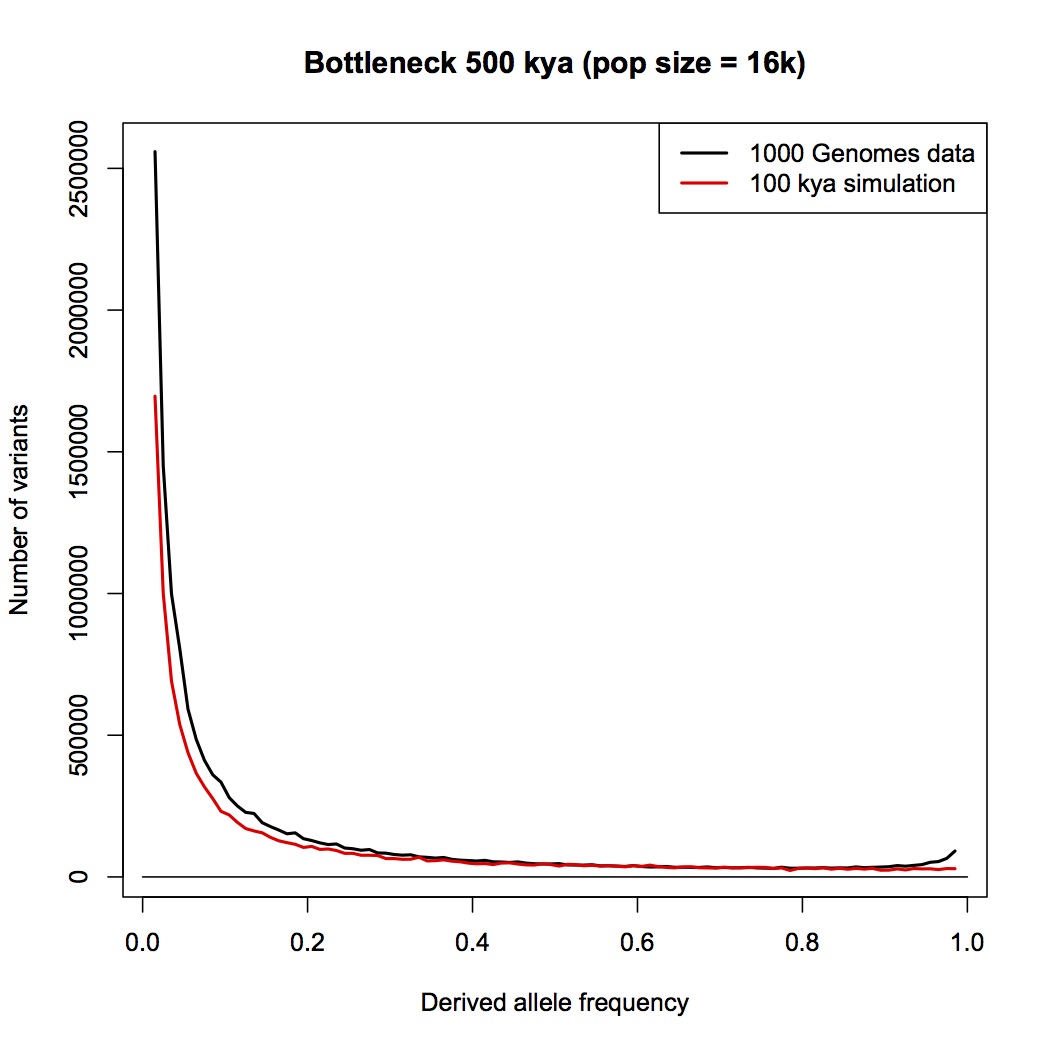
I am sorry Dennis, but I am not persuaded that this passage in your book is a summary statement that includes the PSMC method. With all due respects to you as author, a plain reading of your chapter, as I have spelt out in detail above, is that this passage refers to an allele counting method that you then later compare the LD and PSMC approaches with. You make a point in your chapter that allele counts, LD and PSMC independently give close to the same result - a population size of 10,000 individuals.
Furthermore, in your Part 1 response blog (which we are discussing here) you make a big point that heterozygosity is little affected by bottlenecks but allele counts are. You go to great length to explain why allele counts are a good way of detecting bottlenecks. You repeat the claim that the allele counting method indicates that human population sizes have never dropped below 10,000.
But now you seem to be saying to me that allele counting methods are not actually specifically included in your chapter: that the passage about the allele counting method is actually a summary about all methods that use alleles in some way, including PSMC (which does not count alleles, and does not “select a few genes”). Despite my repeated requests, you have not given me any reference or citation, or a description of an analysis that you or someone else has done, where human effective population sizes have been estimated by an allele counting method.
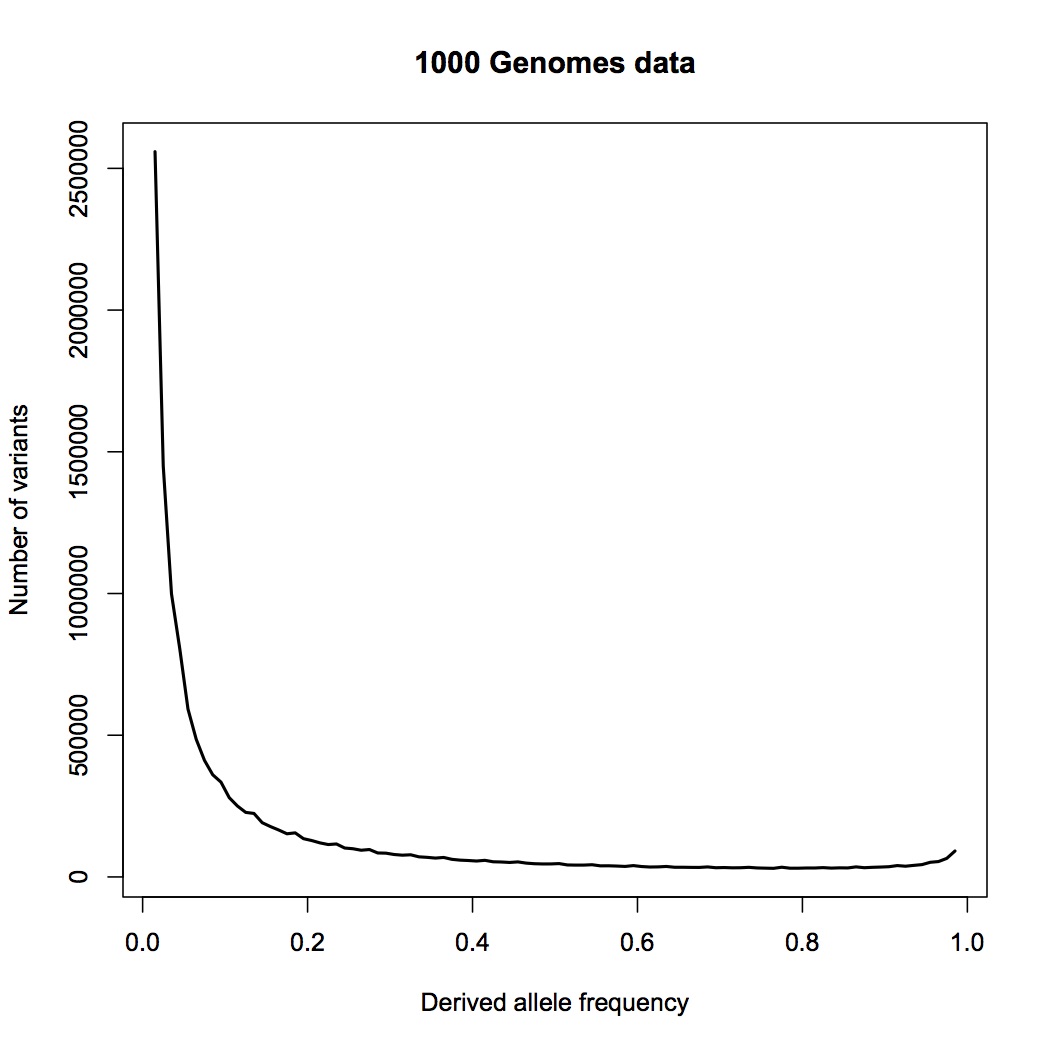
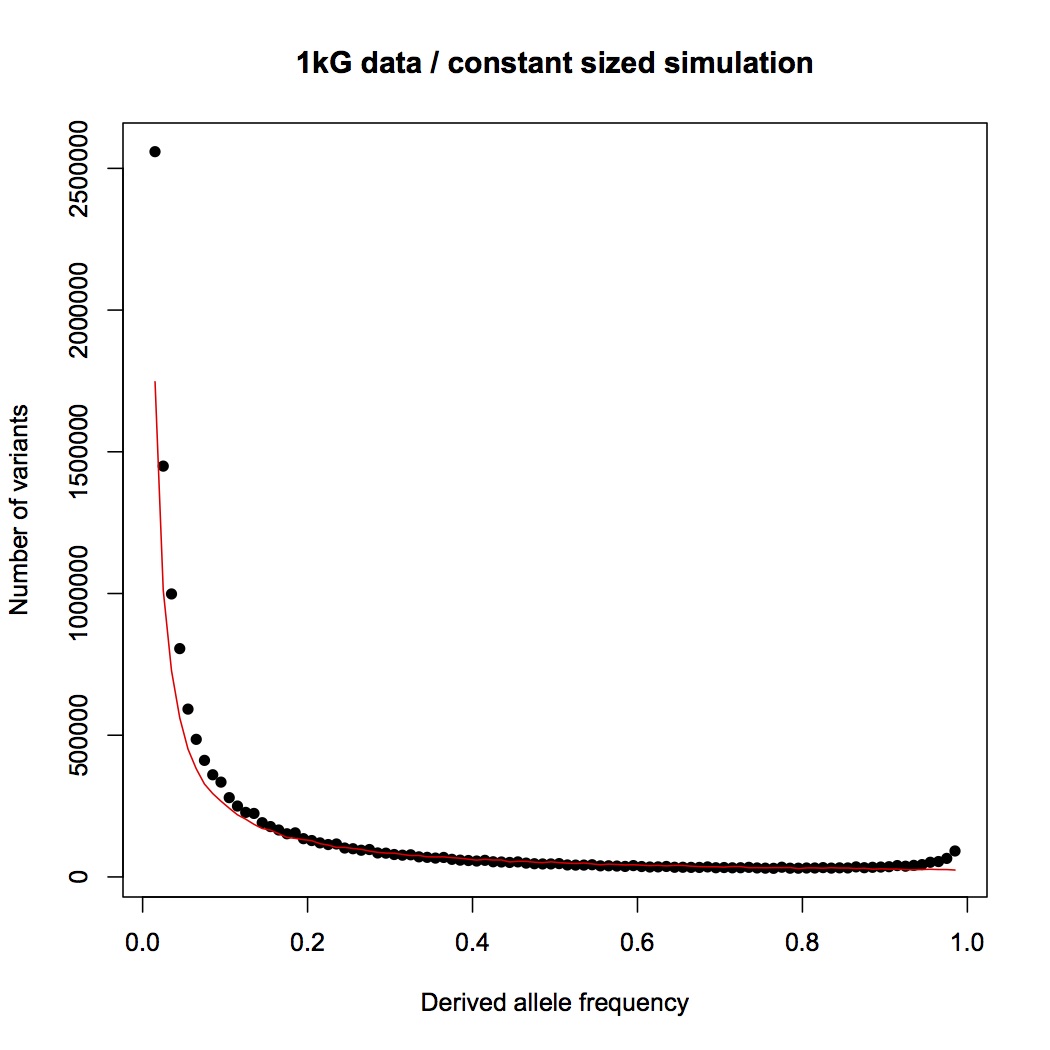
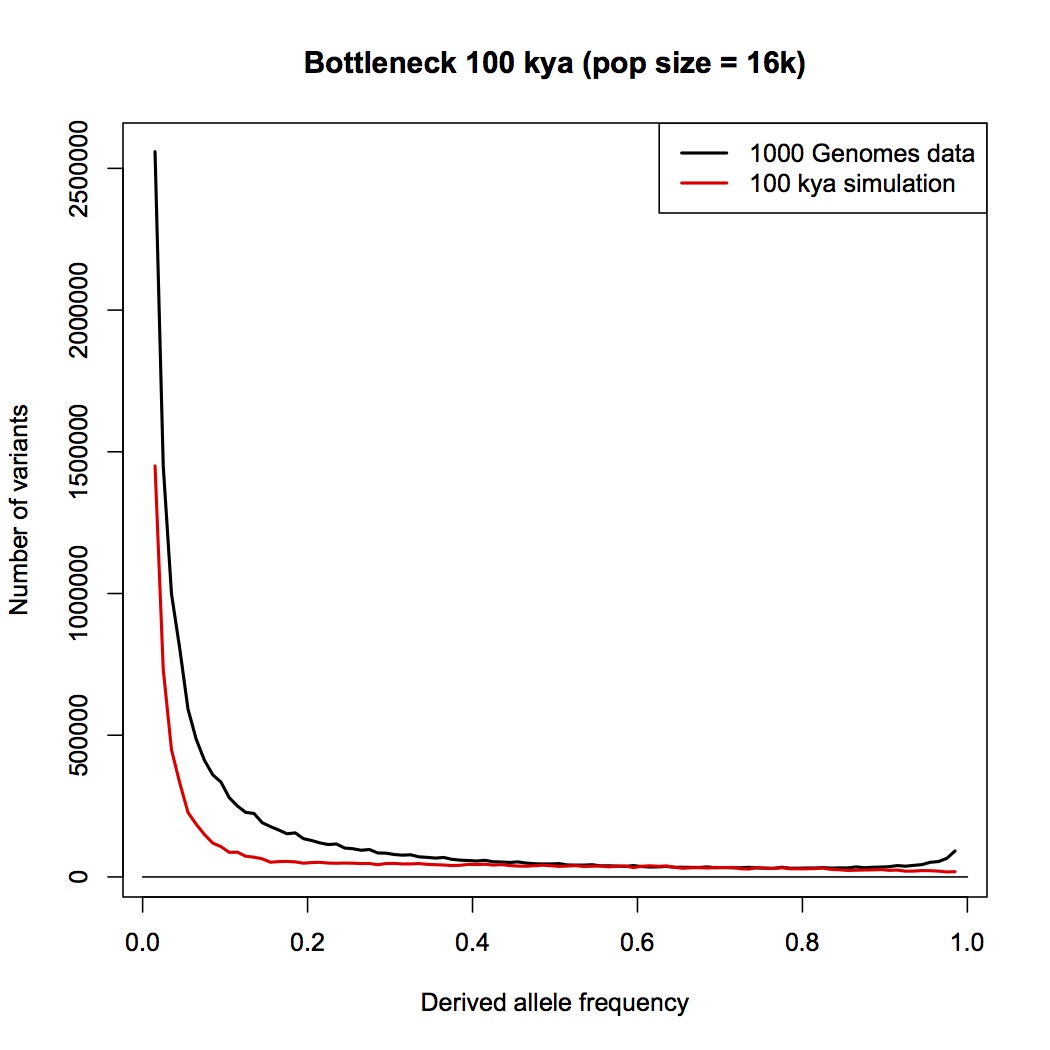
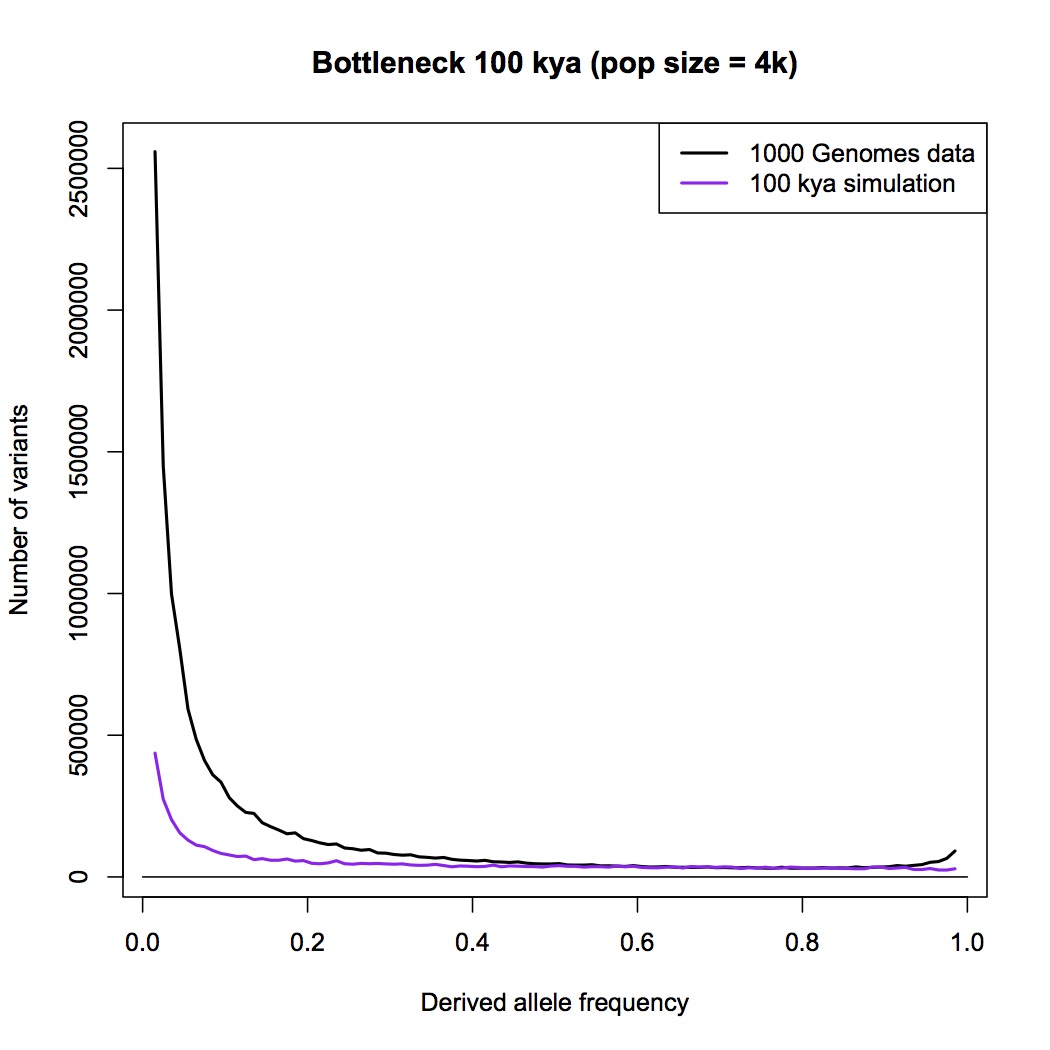
Instead, you are pointing me to the 1000 genomes paper. This is a wonderful paper that I have often referred my students to, and I do not doubt for a moment that the 1000 genomes project provides the raw data necessary for an analysis based on allele counts, but as far as I can see, the authors have not done such an analysis.
If you are not able to give me a citation that includes use of an allele counting method, why did you spend such a large proportion of your Part I blog explaining why the allele counting method is such a good way of detecting bottlenecks? Why do you mention allele counting methods in your book?
I have to admit, I am bemused by this. I think that the allele counting method is one of the best methods available for detecting bottlenecks, and I think it is the biggest challenge to the bottleneck of two hypothesis. I think there is a really interesting discussion to be had here. It has come as a genuine surprise to me that you are not pointing me to a calculation, or a paper, or a textbook, or something else that clearly explains the derivation of a 10,000 effective population size figure.
We seem to have reached an impasse on this point. I will have to let others read through your book chapter and your blog above, and reach their own conclusions.