That’s an assumption.
It is also unclear how @glipsnort’s experiment is meant to demonstrate mutations are ‘random’ in the undirected sense that you mean. Sure, if biochemical distribution he derived is a sufficient statistic for all the mutations, then I can buy the mutations are undirected from what he presented. However, there is a very large amount of other patterning, especially the distance based correlation, that is inexplicable (to me!) from an unguided perspective, and is certainly not accounted for by the biochemical distribution.
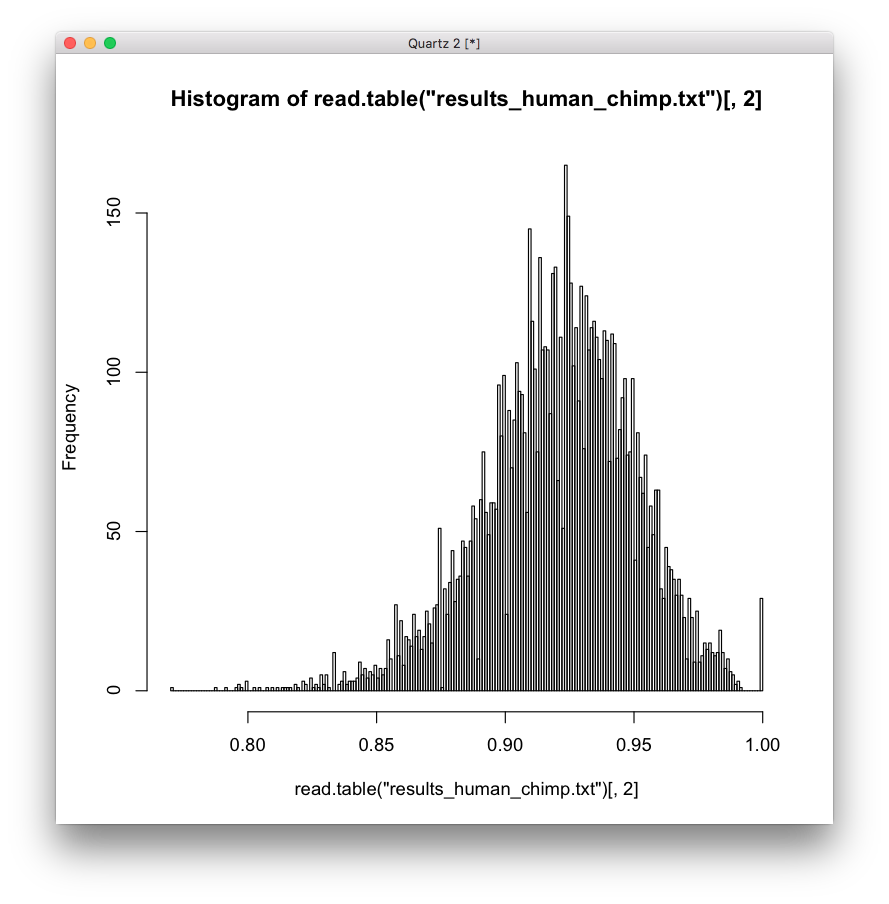
For example, here is a histogram of the human/chimp comparison, which should be the most random, and it seems to be multi-modal, highly skewed, with a possible frequency distribution, definitely not a normal distribution. And if I run a test for normality on it, I get a very small p-value = 3.952e-16.
This histogram for human cow is even weirder, after I increased the number of bins to 600.
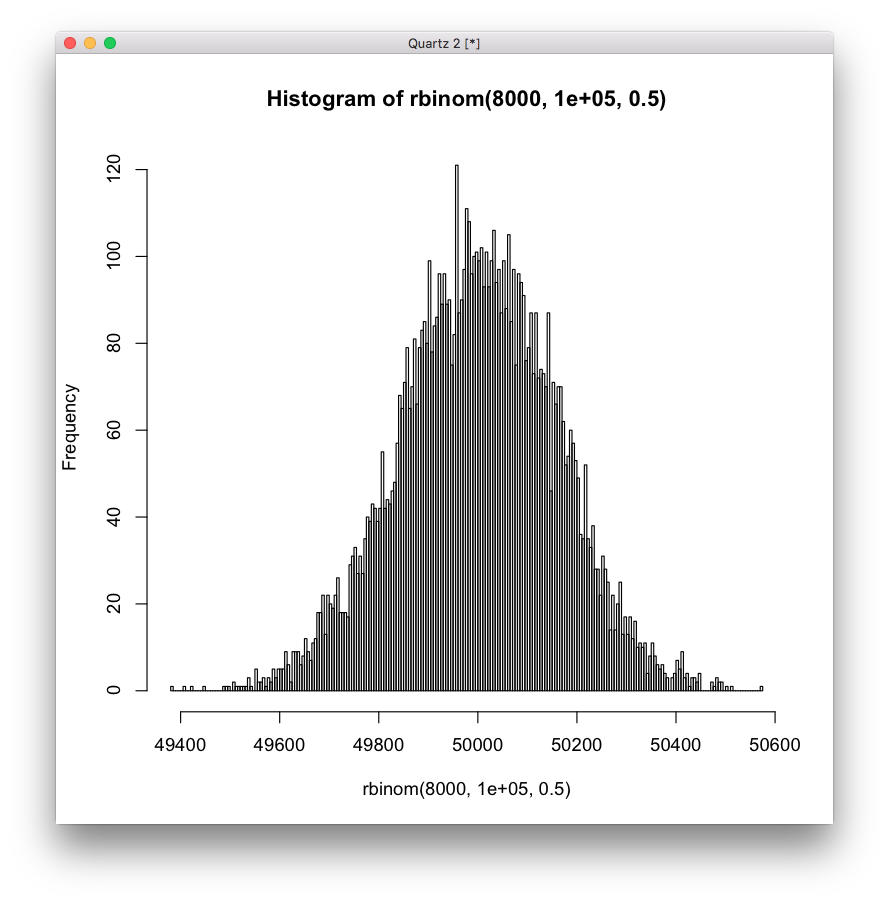
As a comparison, here is a histogram from sampling a normal distribution the same number of 8000 samples, which a test for normality gives p-value = 0.5557.
I guess you can chalk all patterning up to ‘natural selection’ but then what can’t natural selection do? It becomes yet another ‘of the gaps’ entity that we use to file anything weird under ‘misc’ 
And if we want to put constraints on natural selection, it can only select for what is immediately beneficial. If in order to get benefit we need multiple, coordinated, distant mutations to occur, it is very unclear how natural selection can help, since a very improbable event has to occur for natural selection to have any effect.
Here’s one section that pops out to me in the paper:
On the basis of this analysis, we estimate that the human and chimpanzee genomes each contain 40–45 Mb of species-specific euchromatic sequence, and the indel differences between the genomes thus total ∼90 Mb. This difference corresponds to ∼3% of both genomes and dwarfs the 1.23% difference resulting from nucleotide substitutions; this confirms and extends several recent studies63,64,65,66,67. Of course, the number of indel events is far fewer than the number of substitution events (∼5 million compared with ∼35 million, respectively).
This sounds bigger than the 1% number that is quoted, more like a total of 4% difference between chimp and human. And big insertions of new genetic material (if I understand correctly) seems very mysterious from an evolution point of view. I’m guessing the assumption is this inserted/deleted material was present in the ancestor, and different parts were deleted in humans and chimps.
Another thing, the way the genomes were compared it seems they were chopped into 1Mb pieces, and then pieces were compared. Doesn’t sound too bad, but it is less straightforward than if the genomes were lined up side by side and directly compared.
Additionally, the general use of edit distance type comparisons to find the SNPs is also a bit tricky. I can generate two random strings, and discard everything that doesn’t match as deletion/addition events, and the two strings will be considered perfect matches. Not quite what is done here, BLAST type algorithms try to find minimum edit alignments, but the discarding of portions that don’t match does add ambiguity. This ambiguity is absent from the popular caricature that scientists are lying both genomes down side by side and comparing them base pair by base pair.
Finally, the method of assembling the genome using a human reference is still yet another source of ambiguity. When sequencing, enormous numbers of reads are generated with some degree of error, and reads are not very long, just a few hundred base pairs long. With enough short erroneous reads you can match any string.
In general, with sequencing, assembly, and comparison there appear to be a sizeable number of opportunities for overfitting, so I think the human/chimp 1-2% comparison number needs to be taken with a generous pinch of salt.