I am one of the readers who wants to see the evidence for a bottleneck of two. If I have missed it in all the posts, could you please link to it? Are there any scientists who argue for a bottleneck of two any time in the last 100,000 years?
As I said before, I have only one question for this discussion. Does your understanding of the data support the idea of an Adam and Eve who had no ancestors at all (neither human nor pre-human), as the universal progenitors of every human who has ever lived, with no humans descending from any parallel humans or pre-humans, approximately 6,000 years ago? That’s all that matters to me.
While we’re waiting on Dennis’ reply, I’m hoping you can bring a little more clarity in your main critique with respect to the studies he referenced for you.
For what it’s worth, I can give you my own impression of what may be happening here by way of analogy. Let’s say we have a YEC criticize the available peer-reviewed literature for not testing his “hypothesis” of an ice canopy. While the mainstream scientist may provide you with references till they’re blue in the face on paleoclimatology, physics, cosmology, etc., you continue to object that your particular “hypothesis” of an ice canopy has not been explicitly tested and rejected and so all this bravado from scientists on their confidence of what the earth looked like in the ancient past is faulty. So, as this mainstream scientist just cannot dig up a laser-focused rebuttal to your particular arbitrary model, they simply run calculations for you demonstrating the implausibility of your claim. Which you then take to be a weakness of the existing literature. Why run the numbers if the available studies already refute it you say?
I think (or hope) we can all see the absurdity of such an approach. And while I’m getting the sense that this MAY be what is happening here with your critique on Dennis, I don’t know it. So I’m hoping you can clear this up for me. What are your main issues with how he is or is not engaging the available literature? As succinctly and clearly as you can sum them up? I look forward to your thoughts.
Agreed, I haven’t even seen anything like this at AIG or ICR. Granted, I don’t read exhaustively at either site, but such a claim would’ve likely caught my attention.
Organizations like AIG and ICR have made a kind of hamfisted attempt to do this with lots of fuzzy talk about Mitochondrial Eve, and AIG has made its own claim about a genetic bottleneck. This recent genetic bottleneck argument is obviously being promoted by the Discovery Institute. However, CMI is following the current discussion at Biologos.
In order to “see” what I’m getting at, you would have to look at the 1000 genomes data and assemble the various haplotypes in their sample from the region in question. There are more in the database now, obviously, than when the paper was published. This is part of what I was saying when I said that some of what you are looking for is just familiarity with published data sets.
This region of the genome is not atypical. It has a haplotype structure like other regions, and scrolling through the data yourself is the best way to convince yourself that it could not descend from just two individuals.
If you want to see how this sort of thing looks, here’s a paper (PDF) that does this for a limited data set for the entirety of human chromosome 21. Look at figure 2 - those are 20 individual chromosomes from their sample (African, Asian, Caucasian). Note well that this is an early paper with a limited data set, but there are more than four common haplotypes even in this very limited data set. Note also how closely linked these SNP sites are. The problem for a 2-person bottleneck hypothesis only gets worse as you add in the reams of data we’ve added since.
Richard’s suggestion that all common variation was packed into two founding individuals and then recombined afterwards into the patterns we see today is actually found in the YEC literature. (Yes, I spend too much time on these things - sigh). Robert Carter is an example of a YEC who tries to get around the bottleneck/founding with 2 problem this way. The challenge for YECs (and Richard) is explaining the patterns we see in the present day with reasonable recombination events. There is also the issue of drift to intermediate frequencies, because small haplotype blocks are effectively “alleles” that can increase or decrease in frequency like individual variants. Although I think Carter starts with a population of one, since Eve is a clone of Adam (presumably with a subtracted Y and a doubled X). In this case, you have to deal with only two original chromosomes, not four.
Why would @DennisVenema need to describe separately what @glipsnort was modelling, if Steve himself already explained very clearly what he was modelling? What purpose does that serve, other than “testing” Dennis? It appears to be an unnecessary demand that does not further the conversation.
And it points out the problems with a very literal interpretation. The genetic makeup of Eve. The origin of Eve’s biomass (what does a rib weigh? a pound, depending on which rib?). The genetics of the offspring. The problem of creation of mankind on the 6th day vs Genesis 2 story. To take it as addressing science places you in a strange position if you try to explain it all. I do not mean to sidetrack the conversation, but these issues are what are going on in the background in my mind when these sorts of discussions come up. If further discussion along these lines is desired, we can move to another post to maintain the integrity of this discussion.
In some ways it is relevant to the conversation… Richard has been asking about a bottleneck to two individuals, but perhaps a bottleneck to one is what really should be discussed here. In that case we’d be down to only two starting chromosomes.
Would not a better start to a good discussion be to analyze the relevant data yourself to see if the data is suggest a bottleneck of two before you enter the public discussion and make many demands of Dennis?
I am disappointed in your failure to point anyone to citations that suggest a bottleneck of two. Why does Dennis have so much responsibility, and you seem to have none?
Wakeley’s results do not apply to your scenario. You do not have enough time to attain a “fairly stationary state”. As a result, I don’t see how you can draw any relevant conclusions from it.
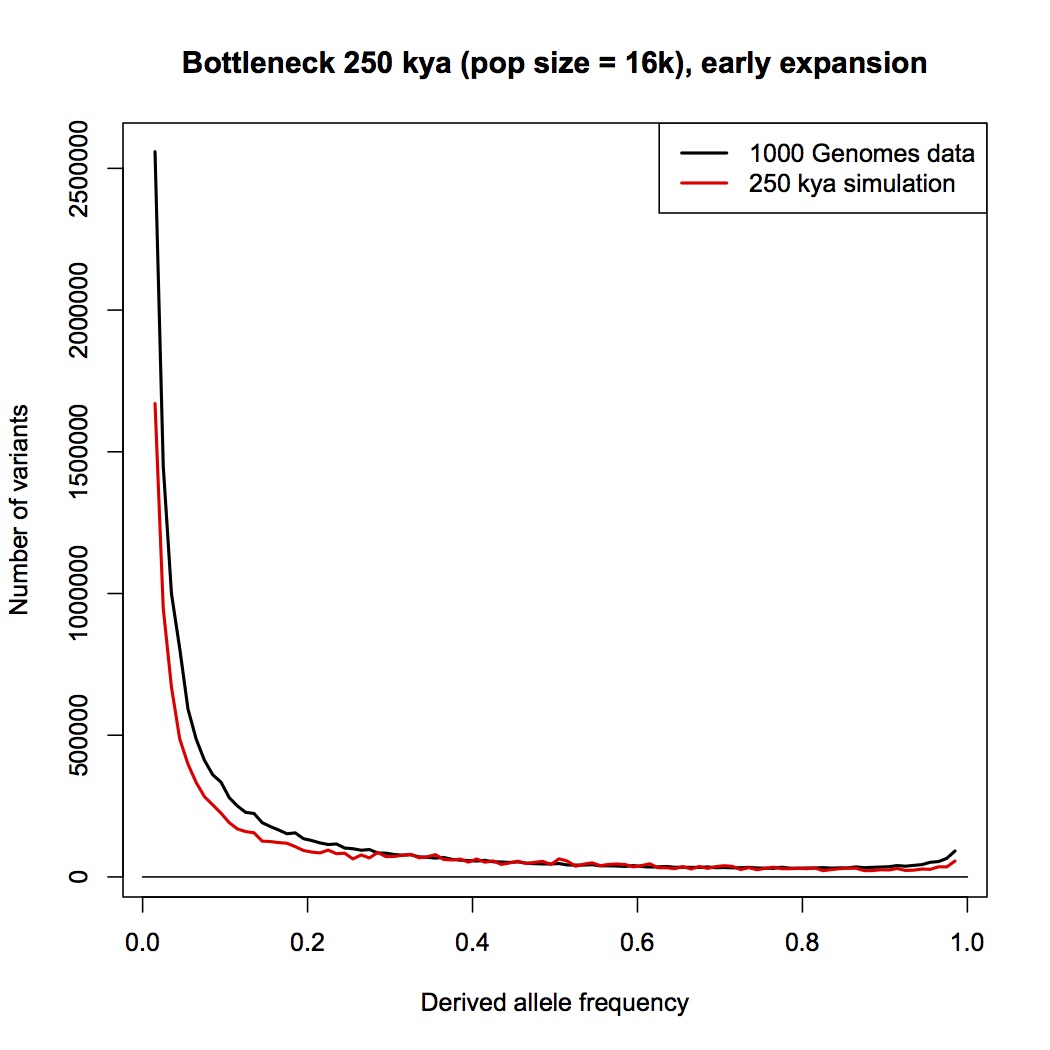
I don’t see a sudden drop at 0.05. I only see the drop at 0.10.
But the problem isn’t to find intermediate frequency alleles. You can do that just by increasing the ancestral population size to make it as large as you like. The problem is getting the frequency distribution to look like 1/frequency. How does population structure help? I don’t see why that would do anything but give you slower and more granular drift of the ancestral frequencies.
A more modest bottleneck that that.
Then it won’t fit in the 60% - 70% range. What you can do is have the ancestral population biased toward lower frequency alleles, so that more come through that bottleneck at 25% than under the constant-sized model. I ran one such scenario last week. Here’s another, which is the best I can up with; it has a rapidly expanding population prior to the bottleneck:
It does give a better fit above 30% frequency, but it still has two distinct regimes, reflecting a change in population dynamics. It now looks pretty much like a major population expansion happened 250,000 years ago. It also requires an implausibly large ancestral population. I’m still using the trick of scaling up the ancestral size to fit the 60-70 window, (even though it’s no longer valid, since the ancestral population wasn’t stationary). Doing so yields an effective population size prior to the bottleneck of 1.4 million. Modeling it properly would make it even bigger.
(I’ve also started modeling incorrect ancestral allele assignments, since we’re looking at the higher part of the spectrum now.)
This has been taking up quite a bit of my time, and I really can’t afford to keep doing it much longer. In my experience, the number of people who care about this specific scenario – which involves jettisoning a fair bit of the actual Genesis story, after all – and who are likely to care about pop gen simulations is pretty small.
Hi Dennis,
Regarding the 10,000bp region in the Zhao et al paper you say this:
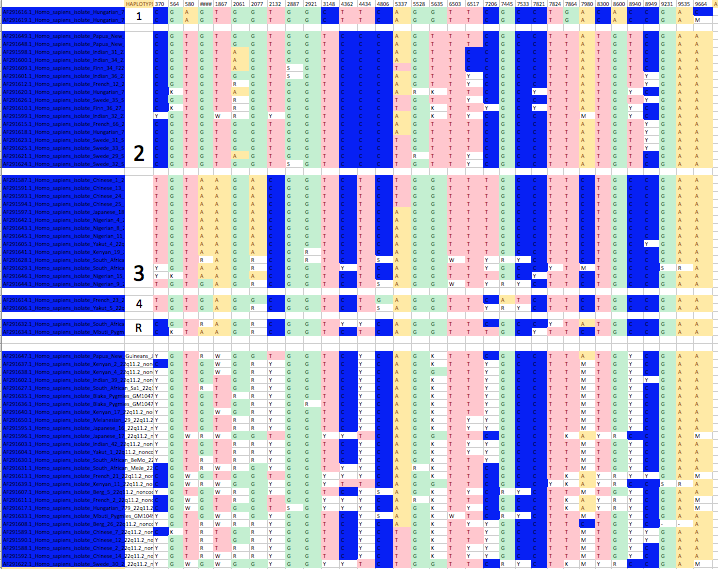
To see if you are correct I have downloaded Zhao et al’s sequences from Genbank, aligned them, and taken a look at the variation. Here is a very simple portrayal of the variants that are present in more than two individuals.
Each column is a locus with variation (with only the loci where the minor allele is present in more than two individuals being shown). The number at the top of each column is the position within the 10,000bp sequence. Each row is an individual human. I have coloured in calls that are unambiguously A, T, G or C.
Many of the calls in the data are ambiguous, which means that either the individual was heterozygous at that locus, or the sequence data was poor there. Where there are several ambiguous sites in an individual it is not easy to infer the haplotypes (combinations in which the variants are found within the two parental genomes within the individual).
In the figure I have taken the individuals that had few ambiguity codes, and divided their haplotypes into four groups, where each halplotype group contains differences of up to three mutations. I have labelled these in the second column, and put a spacing row between them. I have also labelled a couple of sequences as R in the second column, as these may be recombinants. Then at the bottom I have placed individuals that have lots of ambiguous bases, making their haplotypes hard to call.
I think it is fairly clear to the eye that the data present can be divided fairly easily into four groups, that could correspond to small variations on four ancestral haplotypes. Given the similarity of them all, the number of ancestral haplotypes could in fact be lower.
This is a very rough and ready analysis that I did partly on the London Underground on my way home from work this evening. I can follow up in more detail if you wish. As a preliminary analysis, I think it supports my point quite nicely.
I think there is uniform and general agreement that Dr. Schaffner has been through this thoroughly. And he has paid his debt to all those people who have rejected, and will continue to reject, his findings no matter what those findings are.
However, if you still feel the keen and heartfelt pain of those who really want someone to pin his or her reputation and credibility to the unfathomable “bottleneck of a mating pair 6000 years ago”, it would seem you are the perfect candidate.
You are an academic.
You really care about their pain.
And you seem to have an endless list of permutations that need testing.
Now that you’ve looked at that particular data set and tried to “bin” things into four starting chromosomes, you can start to see that there are some differences between individuals in your bins, and that those differences are shared by other individuals within your bins.
Are those differences the result of de novo mutations, or recombination from the starting four haplotypes, then? And is there time for those variants, once they are produced by rare events, to drift to an intermediate frequency?
For example, in your “bin 2” there are three individuals that share an A where others have a G, and those same three individuals also share a C where no one else does. Why is that not another haplotype in a separate bin in your thinking?
It’s worth noting that if God is able to instantaneously organize literal dust particles into the entire human genome in order to make Adam, there’s nothing saying that when God took the rib from Adam, he couldn’t have miraculously generated a second genome for Eve in the time between the rib removal and Eve’s full development.
Of course, this would be an interpolation, but hey, so are most of the details when you try to force a scientific worldview onto a Biblical text. Dino-riding, anyone?
Lastly, it strikes me that if Eve really was a genetic clone of Adam, she would not have been judged to be a suitable helper for Adam. To the extent that my wife and I make a good team, it’s because our strengths and weaknesses even out. If we were genetically identical (minus a Y, plus an X), Lord help us…