Grammatical ambiguity aside, I’m still hung up on the idea that species boundaries are subjective, so all the M&Ms in the bag are brown. Whether “only two” of them are subjectively identified as greenish brown instead of reddish brown is beside the point to me if we can count them and see there are clearly not “only two” M&Ms in the bag. It doesn’t matter what color the M&Ms are. If there was no bottleneck of two individuals in the population, that is the relevant piece of information.
4 Likes
Well said! Context is everything.
1 Like
I would put long-term Ne at probably around 20,000, based on π = 0.1% and µ=1.25e-8. (Note: that’s 20,000 diploid individuals, not 20,000 chromosomes). I used 10,000 and 20,000 generations in most of my simulations, corresponding to 2N/4 and 2N/2. 2N translates to about 1 million years (or somewhat more if the generation time is a little longer), and a TMRCA of 2 million years, since TMRCA is 4N for an ideal population.
Does that make sense?
Sorry for my very spotty attention to this thread, but life has been a little complicated lately.
2 Likes
Make a flow chart or something… because your ability to use words “to make things clear” seems to have become attenuated by the bubbly enthusiasm you have developed regarding this topic.
If you intentionally strip out the time frame from your assertions, you are making an error.
If you carefully explain that the “Bottleneck of One Pair” is something that could have occurred 200,000 years ago… then I am fine with that.
But when you use confusing terminology “before 10 kya” (huh? which direction?!)… and the like, you are just asking for trouble - - especially when you combine it with a double negative and/or a subjunctive verb.
George (George #2, the other George)
1 Like
Thank you for putting a time frame in that sentence… I believe you have single-handedly saved my sanity!
Lynn, I should let you know that I’m a single man again … ![]()
1 Like
Dear all,
I have now started to respond to @DennisVenema Part 2 blog reply to me. I have posted Part 1 of my response here:
And placed it on the Biologos forum here:
best wishes
Richard
1 Like
Thanks @RichardBuggs, looking forward to reading it. Though, coalescence analysis is entirely encoded within ancestral recombination graphs. I’m not sure how relevant it is. The fact that TMR4A is at 500 kya is nearly at the point at which we have demonstrated that coalescence analysis cannot detect a single generation bottleneck of a single couple. I’m not sure his “part 2” requires a response.
Okay, got a chance to read it. I’m not sure your critique is correct. Can you help me understand? You point out two mistakes in this claim by @DennisVenema:
his means that about 25% of the time, heterozygosity is lost, and that only one allele remains in the population for a given gene. If only one allele is present, then this is a coalescence point for that gene: going forward, we will have to wait for mutations to produce new alleles, and those new alleles will coalesce back to their single ancestral allele that survived the bottleneck. In the future, as new alleles are produced from the surviving allele through mutation, the new alleles will all coalesce within a few generations of the bottleneck. Their TMRCA values will thus be almost identical… Coalescent-based methods are thus an excellent way to detect bottlenecks—even really brief ones, if they are severe enough. Even a brief, severe bottleneck will still greatly increase the chances of alleles being lost, and the telltale signature of numerous genes that coalesce within a short time frame.
To clarify here, Ne (or effective population size) is just another unit conversion. It is the reciprocal of the coalescence rate as a function of time. Coalescence are the points in the tree where a merger happens, and they are normalized appropriately by the Kingman term. We just look at when they are, binning by time. This is the coalescence rate. One divided by that is Ne. That is why the TMRCA is relevant (of a subset of the alleles), as that is how the date is determined.
You say his wrong in this claim. I agree, but for a different reason than you. You write (dealing with each point differently)…
I think that Dr Venema is wrong in making this claim. Let me explain why. I think that he is making at least two mistakes here.
(1) In calculations that show that 75% of heterozygosity would be maintained after a bottleneck, the level of heterozygosity before the bottleneck is “known”. But coalescent models run backwards in time, and we can only “see” those lineages that survive the bottleneck. Thus we cannot directly know how many alleles were lost via sampling at the bottleneck. The loss of alleles via the sampling effect of the bottleneck will not show up as coalescence events in a coalescence model. These are two separate effects of a bottleneck.
First off there is a large conceptual difference between coalescence and heterozygosity. A very high amount of coalescence can take place, even as 75% heterozygosity is maintained. These are just different things.
Moreover, the loss of alleles can show up as coalescence events. However, and this the critical point, our ability to detect them is very tightly dependent on the number of lineages entering (in backward time) the bottleneck. If there is only one surviving lineage, there will be zero coalescents, our ability to detect is zero. If there are 50 lineages, there will be a very high amount of coalescence (at least 46 lineages will coalesce), and we will almost certainly detect it. If there is 4 lineages, it seems that we would not detect it. To close the loop, the fact that there might by 75% heterozygosity after a bottleneck tells us nothing about how many lineages are coalescing at this point in time. These are different things, and are entirely separable.
Moreover, and this is a critical point. Coalescence analysis CAN detect bottlenecks, but only if there is sufficient surviving lineages to cause a spike in coalescence at the bottleneck. So @DennisVenema appears to be in error, in that he did not understand the or explain the lineage number dependence on coalescences when he wrote this. However, he is correct in his claim that coalescence can detect bottlenecks, if we limit ourselves to very recent timepoints. However, in the distant past, not so much.
(2) Dennis is assuming that if only one allele is present in a population, then that allele has coalesced. This is a misunderstanding of coalescent theory. In coalescent theory, two gene lineages only coalesce when they reach a single copy in a single genome within a population. This means that if only one allele is present at a particular locus in a bottleneck of two, we know for sure that this allele has NOT coalesced, as it is present in four genomes (two in each person). It must therefore coalesce before the bottleneck. If the ancestral population is large, that coalescence will be a long time before the bottleneck.
I do not think this is his assumption. Coalescence does NOT make any statements about the number of alleles at a given time in history. Rather it make a statement about the number of lineages that survive to this day by direct descent. That is all that is modeled in coalescence theory. It does not presume that all alleles collapse to a single allele at coalescence, just that two alleles (potentially of many) collapse to one.
So once again, a lot of coalescence can take place, even when there is heterozygosity. Let’s look at your figures to make that clear.
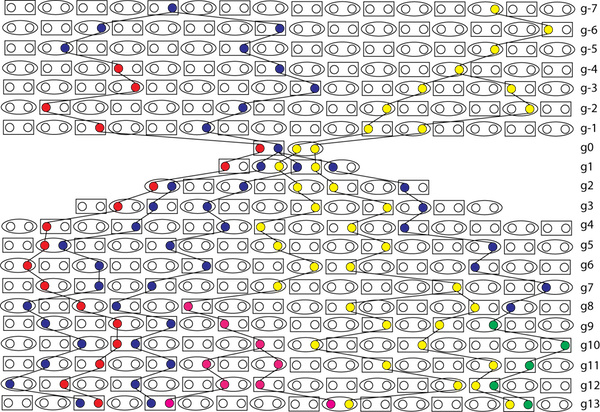
So, here, we see that the conditions put forward are not met. There is more than one lineage going through the bottleneck. The heterozygosity is high.
What about coalescence? Well, in that figure, there are THREE coalescence events between g0 and g1, all in the blue lineages. I cannot tell for sure (it gives me a headache to look to closely) but that seems to be the maximum number of coalescence in any generation. If we had more lineages going in there, we would have seen more coalescence. In fact we are guaranteed that there at least L - 4 coalescents through a single couple bottleneck if there are L lineages going into it (reverse time).
In this case, there was SIX lineages entering, and THREE coalesced. Notice how this nothing to do with heterozygosity. It is, rather, about reduction in number of alleles. It is, also, tightly influenced by the NUMBER of surviving alleles that are still in the population right after (time forward) the bottleneck.
It seems, however, @RichardBuggs you are confusing the several coalescence that appear with “THE coalescent,” which you identify in this figure:
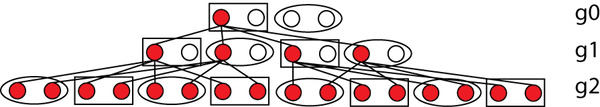
There is not one coalescent at g0, but FOUR coalescents. This is a critical point. Really all merge points in a tree are coalescents, not just the top one.
We can calculate the chances, because normally we would expect there to be a 0.5 chance of a parent passing a particular gene copy to their children. So the chances of what we see in generation g1 above are: 0.5 x 0.5 x 0.5 x 0.5 = 0.54 = 0.0625. The chances of what we see in generation g2, given what we see in generation g1, are 0.516 = 0.0000153. The overall probability of this is 0.520= 0.000000954.
As we have four starting lineages at the bottleneck, we need to multiply by four, to find the overall chance of having coalescence to one lineage at the bottleneck. This gives us 0.00000381. So all in all, we expect coalescence to a single lineage 0.000381% of the time. Not 25% of the time.
This turns out not to be quite correct. Coalescence analysis does not deal with this. Privately, I had shared some similar computations, but also came to understand it was in error (notice it is not in public). However, this computations miss exactly what coalescence theory is doing, I missed this too the first time around, It is quite subtle.
Some subtle math errors notwithstanding, what is being computed here might be a reasonable estimate of the allele distribution we would expect after a bottleneck if we were to measure it. However, most of that diversity is going to die out (or be missed) before we get to our specific samples. So it is not really valid. What coalescence tries to do is, instead, reconstruct the history of all direct ancestral sequences of the data in our current day sample. There may be other alleles with the exact same DNA sequences alongside this direct ancestors, but coalescence only models the direct ancestors.
Keep in mind, the number of allelic lineages at different points in time, does not tell us the number of alleles in the population at that point in time. For example, lets say we are 3 mya, where the vast majority of the genome has coalesced to a single allele that survives till today. The population at that time, however, is not all homozygous for that allele. That allele, also, might even be low frequency. Rather, we are just saying none of those other alleles survive for 3 million years to present day.
For that reason, demonstrating heterozygosity is not lost does not really make the case here. Moreover, remember, no more than 4 alleles can pass through the bottleneck. Heterozygosity, however, does not tell us how many alleles there are.
If my calculations are correct (and I stand ready to be corrected if they are not) then Dennis is quite wrong to think that 25% of genes would coalesce to one lineage at a bottleneck of two. Less than 1% would.
Neither your calculations nor @DennisVenema are correct. It turns out that the amount of coalescence is entirely dependent on the number of extant lineages that enter (backwards time) the bottleneck. The more lineages the more coalescence, the fewer lineages the fewer coalescence. Also his application of the Kingman coalescent to compute 25% is just incorrect. It is wrong.
One final point about a major conceptual error. Read this statement by @DennisVenema:
Coalescent-based methods are thus an excellent way to detect bottlenecks—even really brief ones, if they are severe enough. Even a brief, severe bottleneck will still greatly increase the chances of alleles being lost, and the telltale signature of numerous genes that coalesce within a short time frame.
This is false. For all the reasons we discussed, but for one additional reason. “Severity” of a bottleneck includes two things: (1) the size of the bottleneck population AND (2) the number of generations in the bottleneck. Severe bottlenecks are a LARGE number of generations, with a SMALL population size. However, a SINGLE generation of a very SMALL population size (e.g. a single couple) is not necessarily a severe bottleneck. That is, remember, because severity is defined along two dimensions. In one dimension it is severe, but in another it is extremely mild.
Of note, I’ve had a chance to interact with some secular population geneticists about this. There is actually quite a bit in the literature that makes this point. It is common for papers here to note the limitations of this approach, that it cannot pick up brief bottlenecks.
That is, in fact, what makes this question so scientifically interesting. Essentially, we are asking if a bottleneck is extremely severe by one dimension, but extremely mild by another, is detectable? No one has tested that before (though I just did!, data not shown), and we are finding out that the answer is “no we cannot detect it much before 500 kya.” It’s no surprise, because this falls out nicely from the math, justifying the use of TMR4A here.
In summary, @RichardBuggs I agree that @DennisVenema was in error, however, I’m not sure your argument is correct either. Can you clarify if I missed something here? I hope I did not misrepresent you. My critique here is based on my best understanding of what you wrote. However, please correct me if I missed something,
1 Like
I think you’re on the right track here, @Swamidass, yes - and I agree I was thinking about the size dimension of the bottleneck without properly appreciating the length aspect. (Though, I still have not seen a reasonable case for why such a bottleneck might have occurred - ~10,000 down to 2 and then back up to ~10,000 with exponential growth - that would be unlike anything we’ve ever noted in nature as far as I am aware).
I also think that the TMR4A is a better way of looking at this overall - it more directly addresses what we’re really interested in.
I’ll be sharing that PSMC modelling with you and @RichardBuggs shortly. Things got busy… it’ll be good to chew over those data together.
1 Like
A tight bottleneck followed by rapid expansion is a well known and observed manner of speciation. It is observed from single couples (and even single individuals) in some animals and many plants. There was a long standing debate about whether this is important in human speciation too, until the trans-species variation argument seemed to incorrectly end it.
The bottleneck can be caused, for example, by genetic interference or by near extinction events. Or by founder events too. Speciation can be a slow gradual process. It can also be rapid, even occurring in a single generation.
Sorry, I see that wasn’t clear. I’m wondering about such an event specifically for hominins - which, we know from other lines of evidence, have been widely dispersed on the planet for the last 1.8 million years at a minimum.
1 Like
From a geographic distribution point of view, it there were at least two, if not three times, that a species (or subspecies) in our lineage arose from a single location, spreading very quickly across the globe to become a cosmopolitan species. It is possible a bottleneck preceded, and perhaps was even casually interrelated, with these events.
There has been several theories put forward in the literature about this specific question. Like I said, this was a long standing debate. There are several speciation mechanisms that requires this, and the question is whether or not these mechanisms of speciation were important ever in our lineage. Some of these mechanisms, it turns out, can be testable. But that is way beyond our scope here. At this point, we can just settle into the fact that this is unknown.
Ultimately, I think it is beyond science to determine if it was precisely a single couple at these bottlenecks (if they exist). However, a single couple may not be implausible in some scenarios. Drift in these contexts is one the most potent and powerful forces of genetic change. The sharper the bottleneck, the more potent a force it might have been.
I hear what you’re saying, but 10,000 down to 2 in a single generation is what @RichardBuggs is proposing. I know of no case of a large mammal (or even any mammal) where anything analogous to this is thought to have happened. Do either of you know of a case? The literature is large, and I may well be missing something.
1 Like
Perhaps Mouflon sheep.
If not, just because we haven’t observed it doesn’t mean it didn’t happen. For example, just because we haven’t observed abiogenesis, doesn’t mean it didn’t happen. It does not appear, to the point, that there is evidence against an ancient bottleneck.
Regarding how “likely” this is, it seems to depend tightly on what one thinks about Scripture. Because we have no evidence on way or the other, people are just going to reiterate their priors. Where is science is silent, theology has legitimate autonomy.
That sounds awfully like a God of the gaps. I don’t think it’s wise to tell people they can be confident basing their faith on things which “might” have happened, which science “hasn’t ruled out yet”.
1 Like
What two or three times do you mean?
2 Likes
I think the example most relevant to this case is the spread of Homo erectus about 2 million years ago.
I see why this model of a very ancient Adam seems valuable to @agauger. We can avoid perceived “problems” of Adam interbreeding with others, and also give an account of “human”-like behavior in non-Homo sapiens.
However, I think @Jon_Garvey’s theological critique from Christology and God’s nature is important.
http://potiphar.jongarvey.co.uk/2018/02/23/the-lord-is-not-slow/
If Adam fell 2 million years ago, that means God waiting 2 million years to redeem us by Jesus. Why would God wait so long? How was His plan of redemption set into motion 2 million years ago? This, to me, are large puzzle that need to be addressed by any ancient Adam model, including the RTB model (with Adam 100 kya) and Figurative Adam models linked to the origin of Homo sapiens too (such as @DennisVenema’s model).
I’ve been looking for a good account of this, but have yet to find one already put forward. I’m stuck on this too, and cannot see quite how to answer this in an any ancient Adam scenario.
These Christological concerns are what pushes me towards a recent genealogical Adam. We could see the rise of civilization as both the Fall into Knowledge of Good and Evil, and God initiating His plan to redeem us through the Incarnation, Death, and Resurrection of Jesus. The testimony of Scripture, for example, seems predicated, at minimum, on written language. This instinct is rooted in my admittedly fallible understanding of God’s nature, but there are legitimate theological questions I’d like to see engaged by ancient Adam advocates (e.g. @DennisVenema and @agauger and @RichardBuggs). In these models…
Why did God not send Jesus soon after the fall?
Why did God not just execute Adam and Eve and start over with a new couple? Or species (in @DennisVenema’s case)?
Why do we not see much evidence of the fall in the distant past, even if we see evidence of human-like behavior? In what way does the Fall radically reshape the world?
In a recent genealogical Adam model, the answers are more accessible. God does send Jesus soon after the fall. The reason God does not execute Adam and start over is because there are people outside the garden too, and allowing Adam to leave sows the seeds for redemption of people outside the garden. Moreover, the Fall does entirely reshape the world as human civilization rises. So we do see a radical transformation of the world, but this is associated more with the fall of Adam than the rise of the human mind.
Of course, maybe there is a way to make sense of this in an ancient Adam scenario. I’d be curious to hear how those who care about that scenario thinking about it. Until then, I see more coherence in @Jon_Garvey’s approach.
You should know by now that I encourage people to place the confidence in the thing that science HAS definitely ruled out: the Resurrection of the Son of God. Those who die do not rise again, yet this is the solid rock on which my faith finds confidence.
http://peacefulscience.org/swamidass-confident-fatih.pdf
As for Adam, there is a big difference between saying science does not know the details, and inferring God’s action because there is a gap in our knowledge. Scripture does not teach of the mechanism of God’s creative work, but some do feel it tells us of Adam. Any one who takes such a position is not resting their faith in science, but looking beyond it.
Of course, those how do not find the Scriptural account trustworth will think differently. Also, there are reasons why some might read that account differently, even if they do find the account trustworthy. This, however, has nothing to do with “God of the Gaps.”
1 Like
There’s not a lot of “human-like behaviour” in Homo at 2 million years ago. A significant issue for ancient Adam models is the conspicuous lack of behaviours we consider indicative of humanity at that time. No art, only simple stone tools, no evidence of intentional burial or religious observance. The picture at 2 million years ago looks nothing like what is described in Genesis.
1 Like
This a matter of great debate within anthropology. You are certainly entitled to your private opinion. Let’s just not equivocate your opinion with the scientific consensus. There is no consensus here. In fact, even the Natural Museum of History seems to be closer to @agauger’s position than yours.
I agree. The same goes for your scenario. It is nothing like what is described in Genesis. Moreover, it raises the same questions as the ancient Adam view, along with several more of its own.