This “heckling” from the sidelines by amateurs detracts from an otherwise interesting discussion. As I read it, @RichardBuggs is questioning the certainty expressed by @DennisVenema (that pop gen modelling is as certain as the earth going around the sun), and he is showing that such modelling includes assumptions that may not be testable.
In any event, the notion of two individuals as “starting” human population, instead of a large number, is only relevant to the biblical account, if we discard virtually all meaning in the account in Genesis - I do not think pop gen modelling can do that.
I am glad to see that you have taken another look at the Zhao et al (2000) paper now and are willing to say this:
So I think (correct me if I am misunderstanding you here) we agree that the authors are not explicitly supporting your case. If you are happy with the comments that I and @swamidass have made on the coalescent analysis at the end of the Zhao et al (2000) paper, suggesting that the data is compatible with a bottleneck in the human lineage, then I am very happy to move on. Please could you tell me which paper you would like me to read next?
I think that to pick up where we were before, it would be helpful if the paper you choose explicitly supports this passage in Adam and the Genome:
“One simple way is to select a few genes and measure how many alleles of that gene are present in present-day humans. Now that the Human Genome Project has been completed and we have sequenced the DNA of thousands of humans, this sort of study can be done simply using a computer. Taking into account the human mutation rate, and the mathematical probability of new mutations spreading in a population or being lost, these methods indicate an ancestral population size for humans right around that 10,000 figure. In fact, to generate the number of alleles we see in the present day from a starting point of just two individuals, one would have to postulate mutation rates far in excess of what we observe for any animal.”
But if you prefer to leave that statement behind now, I would be happy to move onto another method of analysis. If you could tell me which paper you would like me to read next, I will be very happy to give it a close read. I think all of us will be happy to move on to a fresh paper! Thanks again for all the time you are putting into this discussion - I know that all of us have many pressures on our time.
I’m encouraged to see you moving on to Dennis’ stronger evidence, and am glad to see my dialogue with him was useful to you in that regard. I do believe there is an outstanding question from Dennis for you to address. Since I know you’re focusing your efforts in responding to him rather than other readers, I’m confident we’ll see a reply from you very shortly on this:
Since most of the papers in this field use other primates to calibrate mutation rates in specific genome areas, it would be helpful if Richard would answer this straightforward question now before proceeding.
In this region alone, not taking LD into account, the number of variants seems consistent with a bottleneck of our ancestors (I would not use the term “human”) at or before 500,000 years ago. It is possible a more careful analysis will show problems, especially if LD is taken into acoount, but we should not mistake TMRCA in a single autosomal region for a limit on bottleneck time. TMRCA also is not an estimate effective population size.
As long as we are here, the data figure of the region clusters shows that samples are clustering strongly with population, which strongly undercuts the notion that any observable clusters correlate with alleles from a primordial couple, which we would expect to be randomly distributed in geography at this point. This does not necessarily mean it does not fit an ancient bottleneck but it does show another view of the data might weaken the case
In the end it doesn’t matter so much because, as Dennis has repeated, the evidence from other sources are much clearer.
And as such, this paper supports the conclusion I state in Adam and the Genome. If one was to squeeze this variation into the last 200,000 years (or even 300,000 years), one would have to increase the mutation rate to do so.
Yep. Where alleles are found in the present day is also important. If everything tracks back to 2 ancestors at 500,000 years ago, why are patterns of alleles in the places they are? Why did some haplotype blocks end up outside Africa only? Why are others only found in Africa?
In the same sense Richard was not willing to accept Dennis moving on “ad argumentum” from the Zhao et al (2000) paper but wanted something more real, I think Dennis is also asking for Richard’s actual take on common ancestry. I can think of no legitimate scientific reason to refuse to answer this question. Can you? If Richard does not accept what is universally considered established scientific fact within genetics, it would be informative to the discussion. For instance, he may be more inclined to treat things such as mutation rates as assumptions rather than conclusions. Which may color his intuitions on how much freedom he has to play with the parameters to get to a bottleneck of two. I also believe given all the transparency he’s demanded of Dennis, he owes him an answer.
Not a scientific reason, no. But lots of people commenting here have relationships with various organizations and institutions with certain a-scientific commitments, and so I understand and sympathize with a reticence to be completely upfront with one’s personal beliefs and convictions on points that are considered political or controversial in those arenas. People have lost their jobs over things they have said on BioLogos. It should be enough to clarify what the terms of the discussion are.
These aren’t personal beliefs but scientific ones, correct? To my knowledge Dennis is not asking Richard about theological or religious beliefs, but is limiting his questions to scientific ones. Michael Behe has an affiliation with the Discovery Institute, the premier ID association in the US, and he acknowledges his acceptance of common descent without issue to any relationships or standing there. Nor does it look like the Queen Mary University of London where Richard is employed would impose any negative consequences on him over such an acknowledgement.
But to your point, if Richard feels he cannot be open about where he falls on common descent, then at the minimum I hope he has the wherewithal to say so. And hopefully also address whether he would feel similarly constrained to not be able to openly acknowledge whether a bottleneck to two in the past 500,000…or even 200,000 years for that matter is implausible should Dennis effectively make his case in this exchange. We should at the very minimum have a clear expectation of what can be achieved in this conversation.
I can’t imagine why @RichardBuggs would fear for his job by agreeing with the scientific consensus on this point - but you’re correct, there may be other issues at play. I merely want to know if common descent is a given for this discussion. Also, knowing what Richard thinks are reasonable speciation times would also be useful. And yes, there is some irony that I am required to lay out everything in great detail but Richard need not answer even very basic biological questions. I’ll just state for the record that I’ve asked, and if Richard chooses not to reply, then so be it.
Yes they are. They claim that there is no evidence in their data for a severe population bottleneck in non Africans. Even if we ignore the fact that Africans are by implication even less likely to have had such a bottleneck, the authors rule out a sharp bottleneck for non Africans. Even if non Africans were the only people on earth this paper would still count as support for Adam and the Genome.
But, by all means let’s move on and let Zhao (2000) rest in peace.
What next? We could do the Alu paper, but given the foregoing conversation and our discussion of TMRCA and TMR4A values, why don’t we do this one next? I think it follows on nicely from the previous discussion. The authors estimate a TMRCA at over 2MYA, and they also plot out the TMR4A - have a look at figure 3. The TMR4A is over 500,000 years ago. In fact, I count 10 haplotypes at 500,000 years ago in their analysis. This would again support the conclusion I come to in AatG, and it’s in a format that is easy to see.
3 Likes
gbrooks9
(George Brooks, TE (E.volutionary T.heist OR P.rovidentialist))
281
I can understand @RichardBuggs choice of words: “for the purpose of this discussion” … it knits well on the idea of common descent.
But how does he say “and I think speciation, for the purpose of this discussion, can occur in x years…”
I doubt if you will ever get him to phrase an answer that specific!
I think Richard could give some parameters. Such as “I think any bottleneck to two within the last 50,000 years is effectively ruled out by the evidence.” Or 100,000 years, or 200,000 etc. The notion that he cannot (nor can anyone) pinpoint with laser precision a date does not mean you cannot commit to any minimal parameter at all. I would presume Richard could do so if he chooses. If he doesn’t, it’s a safe bet he explicitly chose not to. And transparency and any notion of a free and open discussion on this topic will be the first casualty of such a decision. Ditto if he ducks out of giving an open and transparent answer on common descent. It wouldn’t mean then the conversation isn’t still useful. But we’ll all have to revise downward our expectations for it sadly. As we will have to revise downward the value of any intuitions and judgements of a scientist who can’t be open about what they think scientifically. I hope to avoid that outcome if we can.
gbrooks9
(George Brooks, TE (E.volutionary T.heist OR P.rovidentialist))
283
Maybe, though I hope he avoids such obscurantism. Brings to mind the lawyer trick of: “No, those accusations are absolutely false, I didn’t do those things as they they described them.” Which of course can mean (not by accident), that maybe they did do those things just not precisely as described in every detail. Hopefully we can avoid such parlor tricks here.
gbrooks9
(George Brooks, TE (E.volutionary T.heist OR P.rovidentialist))
285
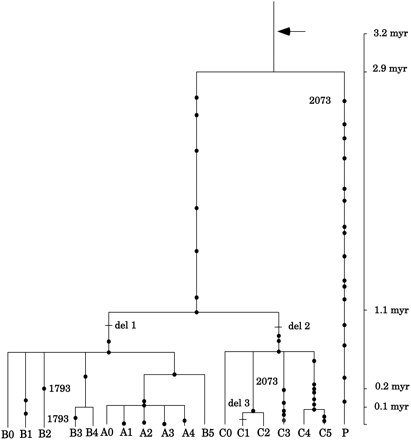
So, in this figure, we see a horizontal cut at 0.5 mya shows 10 branches in the phylogenetic tree.
However, this does not seem to make the case very strongly. @DennisVenema and @RichardBuggs, I had some questions and observations…
It looks like my estimate of (TMRCA / 4 = TMR4A) is holding up. That is encouraging.
At 0.6 mya there is just 3 alleles, and the mutational support for most those splits is low. What do you think is the variance of the date where the transition from 3 to 10 alleles takes place? It seems that a different phylogenetic tree could be easily drawn with the same data that did show 4 or less alleles at 0.5 mya. Even a small variance in mutation rate would do the same thing. @DennisVenema, am I missing something there?
However, it would be much harder to see how there were just 4 alleles, say, at 0.2 mya. @RichardBuggs, what is the minimum time to bottleneck you say is plausible with this data (assuming no miracles)? I know your working hypothesis is 0.5 mya, but does this data rule out anything earlier?
A couple other interesting features is the 1793 and 2073, which are marked because they do not fall into nested clades (either because of recombination or the birthday problem Birthday problem - Wikipedia). I’m sure no one disputes these alleles have the same origin (because they are all human!), so that is a good reminder that we do not expect perfect nested clades in evolutionary processes, and that is not what we see.
I would also point out that the gene involved here (CMAH) is a pseudogene, which means it no longer is producing a functional protien. If we were to say there was a bottleneck here, this would count as some de facto evidence for common descent. As the primordial couple’s genome would include the history of this gene in its genome; a false history if they had been de novo created, and without interbreeding.
If we allow for interbreeding, of course, all bets are off. All this diversity could have been injected by interbreeding events. That, as I understand it, is not germane to this conversation. The question is about a single couple bottleneck, not a single couple origin.
The distinctions between TMRCA and TMR4A do not apply in considering mitochondrial and Y-chromosomal DNA. Does everyone agree that those dates put a lower bound on bottleneck (assuming no miracles and no interbreeding)? That would be at about 150 kya to 200 kya; do you agree @RichardBuggs?
This dataset, however, has all the same problems as Zhao. It is just one region of the genome, and not even that many samples. A better analysis will look at the whole genome, estimate effective population size, and use a larger number of samples. It is in those studies that the evidence appears clearer. And of course, there is still trans-species variation…
He did not duck the question. He answered unequivocally that this conversation presumes common descent. Moreover, as we get deeper into the data, more evidence for common descent is uncovered all the time (e…g the pseudogene).
It seems as if you are asking for him to state what he personally believes about common descent. That is not a relevant question. As should be clear, if we allow for miracles (e.g. mosaic Adam and Eve) none of this analysis is solid any ways. In science, we do not consider such fantastical things, but in one’s personal beliefs we can believe them if we want. As long as that is kept separate from the scientific work, and we are honest about the data, no one cares what we “believe in our heart” about such things.
It just doesn’t matter our personal beliefs, if we are honest with what the data shows.
{kind=link}