Looks like they actually shot the elephant in the room, then dragged it out of the room, and buried it in the yard behind the house.
1 Like
Well, they certainly do “argue that”. And the last sentence says they Propose An Alternative Simulation. Yep, I agree. They are proposing it.
This should be amazing if one is ever produced… even a bad one would be amazing at this point !
1 Like
I don’t see the ‘mosaic’ proposal as viable for recent bottlenecks, without also introducing effectively miraculous genetic recombination and segregation events in subsequent generations across various populations. Perhaps if Adam and Eve were hugely polyploid and ‘mosaic’? I also believe that Buggs suggested a mosaic model when he referenced data and suggested that many alleles in one region of the genome could be traced back to four original regions.
In practice, I think that “two human bottleneck” models generally imply a mosaic starting point in which additional variation is added via later mutations. What the current models suggest is that it requires hundreds of thousands of years to reach a point where statistical uncertainty / noise finally overwhelms the ability to distinguish between 2- vs. multi-thousand population bottlenecks.
1 Like
That is not what mosaic is. It does not require high recombination or mutation rates or an increased genome size. It just means that Adam and Eve had different genomes in each of their gamete progenitors. This cannot arise in normal development, but perhaps God created them this way, with a different biology than the rest of us.
Lest there be any doubt, this is not my view. I am just explain an proposal put forward by others. Explication is not endorsement.
They might as well propose that God miraculously intervened into the population genome, to make people think there was no bottleneck!
3 Likes
I thought the mosaic reference was as you referenced in Wikipedia. Thanks to recombination, I think almost every gamete carries a somewhat different ‘genome’ from the fertilized egg that started the parent.
Thanks for the clarification about “Adamic mosaicism” as gamete ‘frontloading’ with individual, unique and extremely diverse genetics . I understand you don’t endorse it. It’s not clear that Adam and Eve had thousands of children (some accounts suggest roughly 50) or that the current diversity reflected in modern day populations are compatible with the notion. I still don’t see how the Adamic mosaic model is a genetically ‘compatible’ view without additional miracles piled on to get around the issue of bottleneck size in a few thousand years.
Well Eve, I suppose, would have to be a baby making machine. Let’s say she lives 900 years (like Adam) and is having a kid every 2 years; perhaps she has 400 kids, each of whom have two distinct genomes. So that would be 800 genomes. We could also wonder if they were in the Garden longer, and perhaps add to their numbers that way.
Fantastical. Yes. That seems implausible to me, stretching into comical. Poor Eve, consigned to be pregnant and nursing her entire life.
2 Likes
Right? Yet strangely, there are a least a couple major religious organizations that support the notion of frequent childbearing as the desired state. It’s one of the reasons why I’m probably related to about half of the people in southern New Jersey.
No matter how long you live, pregnancy is tough on the body. I know several women with large families, and after a dozen pregnancies, you welcome menopause.
1 Like
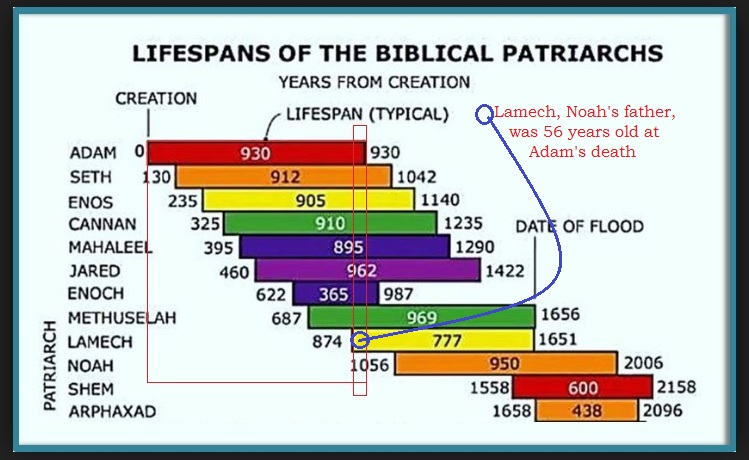
I always think it is a little odd, that with such a long life span, and with these key patriarchs, we never hear stories about Adam and Eve in their latter years … or what a rascal their Great Grandchild Cannan was… Heck, Noah’s father
Lamech was still in his prime of 56 when Adam died at age 930. And we have nothing!
1 Like
Hi Dennis, [quote=“DennisVenema, post:175, topic:37039”]
I’ll be honest that I see this discussion as providing ever-diminishing returns. If you want to see your ideas get traction, it’s time for you to do some modelling.
[/quote]
I am sorry to hear you say that, as I have invested considerable time in this discussion, and I thought that you were seeing it as valuable peer review of your book. What you call “my ideas” are in fact the hypothesis that you took it upon yourself to raise and refute in chapter three of your book. My point is simply that I think your refutation is mistaken and misleading to your readers. My hope is that you feel sufficient responsibility to your readers to make sure that you can deal with my critiques. Your book chapter places a burden of evidence very heavily upon yourself, especially as you claim such a high level of certainty about your view. I have not written a book telling people what science says about Adam and Eve, but you have. I am simply giving you feedback.
When you first pointed me to the Zhao et al paper, there was no hint that you saw it as not being strong evidence. Indeed, it seemed one of the most promising papers to back up the claims you made in your chapter, as it was at least close in time to the human genome project. The fact that you are now suggesting it is weak evidence I take as an encouragement that you are at least taking on board the points I have raised about the Zhao et al paper, and agree with some of my critiques of your attempts to use it to support your case. The only reason why I continue to discuss it is that you are continuing to claim that the final part of it supports your case, even though the authors make no hint of this. I am also interested in continuing to discuss it because it is causing us to discuss data (something that you wanted to do!). I have an inkling that perhaps your very high level of certainty in the case you are making is because you may perhaps be misinterpreting the data at quite a fundamental level, and by digging down to primary data I may be able to identify where you are going wrong - if you are. This is why I am keen to complete our discussions of Zhao et al.
But I am not. As I say, you have proposed the hypothesis, and have also claimed that it is falsfied with a very high level of certainty. I am just querying your falsification[quote=“DennisVenema, post:175, topic:37039”]
you would again have to have drift occur to make these new combinations reasonably frequent such that they would be picked up in Zhao’s sample size. That’s a large number of very rare events, and at least three instances where drift has time to work to take new variants to a reasonable frequency.
You don’t have enough time in your model to make this work. Rare events take a long time to appear, and then drift has to act between each rare event, and that takes a long time too. Multiple rare events interspersed with long times for drift = too much time.
[/quote]
I agree with you that one of my suggested ancestral haploptypes needs three mutations to become reasonably frequent in the population after a bottleneck to explain the data. In fact I said in my initial description of what I had done that I allowed up to three such mutations. [quote=“RichardBuggs, post:143, topic:37039”]
each halplotype group contains differences of up to three mutations
[/quote]
So we agree about that. Regarding time, in Zhao et al, the coalescent analysis for this region gave a mean estimate of time to the most recent common ancestor (MRCA) for this region of 1,356,000 years ago; and the 95% confidence interval was between 712,000 and 2,112,000 years ago. This is assuming a constant effective population size of 10,000. To date a bottleneck of two, we do not need to go back to a single MRCA - we need to go back to four haplotypes in two individuals. As you will know, it is the final coalescence events that take longest time in a coalescent analysis - so much of the time to the MRCA can be after (going backwards ie. before in time) the bottleneck. In addition, if we are testing the hypothesis of a bottleneck of two, followed by rapid population expansion, we clearly do not have a constant effective population size of 10,000. The bottleneck will cause coalescence events to occur more rapidly than they would in a constant-sized population. In fact, we would need to use the multiple-merger coalescent to model that coalescence events in the early generations after the bottleneck. All this would reduce the time taken by the coalescence process. As I have said before, I am not putting forward a particular hypothesis about the timing of when a bottleneck could have occurred - I am just querying your assertion that one has never occurred in the human lineage - but it seems to me that a timeframe of low hundreds of thousands of years would be reasonable for this particular region of the genome, and perhaps lower.
If you are happy to agree that the Zhao et al paper does not support the case you are making in chapter 3 of Adam and the Genome, I am very happy to move on from it and discuss the other papers you have cited.
1 Like
But which methodology are you using to refute @DennisVenema’s refutation?
A. a methdology where you run a plausible set of numbers using plausible assumptions and you get totally different results?
or
B. a methodology where you ask Dennis a hundred questions, and if he ever decides your questions are too farfetched to be entertained, you will portray his lack of response as an indicator that his scenario is flawed?
3 Likes
With respect to Zhao, it’s part of the evidence that you need to account for, yes - even if there is stronger evidence out there. I’m not sure why you’re saying that I think it’s weak. It’s fine. But there is stronger evidence out there, because there are regions of the genome with more diversity. I still don’t think you can shoehorn Zhao’s data into 4 haplotypes, but tell me this: in your scenario, are you proposing that the three mutations were recombined together through crossing over, or that they happened sequentially within one lineage without recombination? What mutation rate are you working with? 1.1x10^(-8), or a higher value? If you’re using recombination, what recombination rate are you using? How quickly does your proposed population expand after the bottleneck, and what Ne does it reach (and is it stable thereafter)?
Even if I was to grant you Zhao, arguendo, just to move the conversation along, how would you handle the haplotype diversity on chromosome 21?
2 Likes
Since I’m (sort of) granting you Zhao (2000), perhaps now is a good time to point out Zhao (2006) - which is basically the same approach, by the same team, to a different 10kb genomic region. Here’s a summary of the haplotype groups they found (using their numbering for the various types):

Once you’re done with the chromosome 21 paper you can look at this one. Again, there are more than 4 haplotypes to account for.
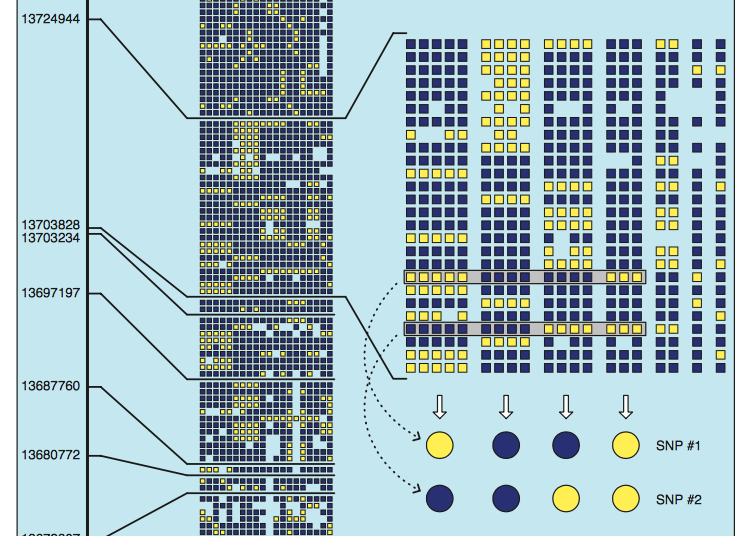
… and just to re-post it so it’s easily at hand, here’s the chromosome 21 paper (PDF). Figure 2 shows the haplotype blocks:
1 Like
Speaking as a reader, I haven’t yet seen you provide an evidence based explanation of why it’s mistaken and misleading. I’ve seen a lot of “what if?” and “maybe”, and claims that the scientific papers on this issue are all wrong, but I haven’t seen you lay out a simple paragraph explanation of your reasons for making this assertion. The longer this continues, the more it looks like Dennis is doing science and you’re doing apologetics. It’s very frustrating because I thought this discussion was going to be completely the opposite way around.
4 Likes
Richard,
Since you’ve put some weight on readers’ impressions on this thread, allow me to give mine on how your latest discussion is coming off.
You asked Dennis, and appropriately noted he was obligated, to produce references to back up the claims put forward in his book. He responded to you with a fairly comprehensive list of citations and some commentary on each. I took this to be him laying out the relevant body of literature. Indeed in my own field (not in the life sciences), I’m well accustomed to citing a body of literature where some studies are run better than others or yield stronger or more reliable results than others and fall across a spectrum of moderate to robust support for the scientific claim being advanced. It would seem the Zhao (2000) paper falls more towards the moderate end of the spectrum, whereas the other papers Dennis directed you to fall more to the robust end. And I think Dennis has been indicating as much.
Yet you react to this in a very strange way for a scientist. So much so it makes me question whether you see your role in all this as one of a scientist instead of, say, an apologist. You seem intent on forcing the papers Dennis references into categories of “weak” or “strong.” With “weak” apparently matching anything that doesn’t pass whatever bar or goal post you’re setting for it. And strong apparently matching whatever does (and one would presume such a bar to be cleared would be truly Herculean in size, if ever even allowing itself to be defined instead of forever ad hoc increasing in height). This forced binary is not scientific. Pushing evidence to extremes to suit one’s agenda. It’s not the sort of thing a scientist would do. It’s the sort of thing apologists do. Quite often in fact.
So I have to tell you Richard, you’re starting to force me into concluding you’re working as an apologist here rather than a scientist. Perhaps you should either revise your approach (I hope), or rather own up to it.
What is more, some of your actions, whether those of a scientist or apologist, are quite galling. You accuse Dennis of being mistaken and misleading, and pontificate about his burden to back up his claims with evidence, while completely avoiding dealing with the studies he himself claims are the most robust he’s put forward thus far. You attempt to critique Steve’s simulations with speculations that a certain population substructure and migration & breeding dynamics, etc. may so dramatically alter the numbers to match a bottleneck of 2 on a relevant timeline. Yet when Steve started to address your speculations, whether by running new simulations or referring you to the relevant literature, you continued to insist that the right mix of these factors could get you to your bottleneck of 2. And then put the burden entirely back on Steve, who had already gone above and beyond by any reasonable measure, to then run the simulations as you want them to be run and you even asked him to go through the extensive effort and time to publish them. All the while you have yet to run a single simulation yourself. To test a single model you propose yourself.
I don’t know how to tell you this Richard, but there’s something very unseemly here. People like Dennis and Steve have been very thorough in their response. And you have not managed to meet their effort. You have studies on your plate you haven’t addressed yet. Ones you explicitly demanded be provided to you. And you’ve sat on them. I suggest before you make a single further demand on Dennis, you read and address these studies. I suggest before you put a single further demand on Steve, you step up and model what you’re proposing. I suggest before you say one further word about somebody else’s burden, you meet yours.
6 Likes
So I’m not the only one who thinks this way. Lest this be deemed inappropriate comment, let’s remember two facts.
-
The only reason why this “two person bottleneck” question is being raised is theology. Scientists aren’t proposing it as an explanation of available data. It is being raised in the context of theology.
-
Dr Buggs has stated explicitly that he considers it theologically useful to see if there is at least a possibility of such a bottleneck, such that it is a matter scientists should investigate (and I agree with that).
I don’t object to using science in the service of apologetics, but when we’re doing so we should be clear that we’re doing so, and not call it something else.
4 Likes
Hi Steve,
Sorry it has taken me a while to respond; my fortnight of teaching has ended now,
I am glad to say and although I now have a backlog with my research group,
I hope I can give this discussion a little more time this week.[quote=“glipsnort, post:169, topic:37039”]
Given enough time, a complicated demographic history can produce all kinds of frequency spectra. In this scenario, though, the core problem is recovering diversity in a short time frame; there isn’t time for complexity introduced by multiple rounds of anything.
[/quote]
I agree, and this is the concern with your model that I am pointing to. I am not sure quite what time scale you are alluding to, as in your original models you covered a wide range of time spans. Are you currently referring to a 200,000 year timespan, or a longer one? Also, as we have already discussed, if the founder couple came from a highly variable population themselves, the issue is less about recovering diversity and more about their alleles at 25% and 50% frequencies drifting downwards.[quote=“glipsnort, post:169, topic:37039”]
There is no mating system in my model. The model assumes a pool of 2N genomes consisting of unlinked markers. For allele frequencies averaged over the whole genome, such details should not matter.
[/quote]
I am not convinced that such details would not matter. I would think that separate male and female sexes, and the complex breeding patterns that this generates in human populations through monogamy, polygamy, sexual selection etc would be bound to have an effect on allele frequencies.
I was thinking of Prado-Martinez et al 2013 Nature. I mentioned this to Dennis in my original email to him: “The average human has 3.1 million single nucleotide variants (SNVs), but the average chimp has 5.7 million (Prado-Martinez et al 2013 Nature). African humans approximately 1.1 heterozygous SNVs in every 1000bp, whereas central chimpanzees have approximately 1.75 (Prado-Martinez et al 2013 Nature).” If, for the sake of the argument, we assumed that the ancestral couple carried similar levels of diversity to chimpanzees, I imagine that would have an appreciable effect on the model. (Note for readers: please do not mis-read me as suggesting that modern day humans evolved directly from chimpanzees - I am just pointing out that present day great apes have higher levels of genetic diversity than humans (despite much smaller current census population sizes) and that it might not be unreasonable to think that our own ancestors had high levels of genetic diversity).[quote=“glipsnort, post:169, topic:37039”]
It would correspond to a spectrum with too many low frequency alleles (relative to a constant-sized population) and some much higher frequency ones, with a relative dearth in between.
[/quote]
But this seems to assume subdivision into just two populations, followed by a merger event. I am suggesting a more complex model which is more likely to reflect the actual population structure: one of multiple sub-populations, multiple migrations – some small, some large – and many mergers and splits. As we have already discussed, this could give smooth allele frequency curve, and the major criticism you have made of this idea is the time available, rather than the ability of complex demographic history to generate “all kinds of frequency spectra”.
I appreciate that it would be a lot of work, and would distract you from your valuable work on malaria. I respect that. I am also not in a position to give much time to this issue, and have to squeeze it into my evenings and weekends. I am grateful though that you are willing to share your code.
However, I have to admit that although I think that your arguments from allele frequency spectra could potentially make a good test of the Adam and Eve bottleneck hypothesis, I would need to see this worked through in considerably more detail before I was fully persuaded that it was an adequate test. I have been reading a bit more widely about site frequency spectra and the factors that can affect them in a few spare hours. In particular I found these recent papers helpful:
Harpak, A., Bhaskar, A., & Pritchard, J. K. (2016). Mutation Rate Variation is a Primary Determinant of the Distribution of Allele Frequencies in Humans. PLoS genetics, 12(12), e1006489.
Ferretti, L., Ledda, A., Wiehe, T., Achaz, G., & Ramos-Onsins, S. E. (2017). Decomposing the site frequency spectrum: the impact of tree topology on neutrality tests. Genetics, 207(1), 229-240.
Koch, E., & Novembre, J. (2017). A Temporal Perspective on the Interplay of Demography and Selection on Deleterious Variation in Humans. G3: Genes, Genomes, Genetics, 7(3), 1027-1037.
Gao, F., & Keinan, A. (2016). Inference of super-exponential human population growth via efficient computation of the site frequency spectrum for generalized models. Genetics, 202(1), 235-245.
These papers have strengthened my view that a wide range of complex demographic, phylogenetic, selective and mutational processes, together with sampling strategies, can influence site frequency spectra, and that I therefore cannot conclude from the models that you have run that a bottleneck of two in the history of the human lineage is not possible. To be convinced I would need to see more complex models run that try to incorporate these factors. I realise that this is beyond the scope of what you wish to do in the context of the present discussion, but I do hope that in the future others may wish to take up the idea.
OK, that is fair enough. I have to admit, I had thought you were doing them to bolster Dennis’s case.
OK, well it’s good to know that you don’t quite have the level of confidence against an ancestral bottleneck as Dennis. What about the existence of Littlefoot? ![]()
Thank you, this is a very helpful explanation of where you are coming from on this question and in this discussion. I hope that Dennis might perhaps send you a copy of his book, given the time you have invested in this discussion. I think that if you had read chapter three of Dennis’ book, and then read my blog on it you might have more sympathy with my critique of Dennis’ case, even if you agree with his final conclusions. I think you might feel as I did when I was flying to Tenerife from London a few months ago, and across the aisle a man said to his family “You can tell we are getting south, as the cabin is getting warmer”. I considered the man to be entirely correct in his conclusion about the location of the plane, but knew that the evidence he had given for it was based on a very partial understanding of physics, climate and aeronautical engineering. If I had had the time, I might have tried to explain to him that whilst I agreed with his conclusion, the reason he had given his children for it was not going to help their science education.
If you read Dennis’ chapter, it would remind you that it is this chapter - and not my blog - that raises the hypothesis of a bottleneck of two, and treats it as a hypothesis that has been raised, tested and disproven with a very high level of certainty.
I respect your view on this as you have a long track record in the analysis of human genetic variation. However, I know that you would not want me as a scientist to resolve this issue with a simple statement from authority. Until I can see the evidence and appropriate analyses clearly laid out in detail and clearly refuting the hypothesis, I can’t see it as decisively disproven.
Given that you do not seem to want to pursue this further due to time constraints, could I finally just ask you these questions (and these can be my final questions to you if you wish): Which published analyses in the literature do you see as most convincingly disproving a bottleneck hypothesis? Do you consider that analyses of the coalescence of different haplotype blocks within the human population could yield decisive tests of the bottleneck hypothesis?
Once again, I would like to say how much I have appreciated your contributions to this discussion. It has certainly moved my understanding of this area forward, and I now have clearer ideas about how the bottleneck of two hypothesis could be tested, which I am very glad about. I am sorry that this has not been as decisive as we might have hoped, but I certainly have learned a great deal. I hope in the future we will see some conclusive testing that can put this issue to rest.
I wonder if we would all concede virtually all your points if you simply declared that even you agree it would take more than 100,000 years in reference to this part of the genome?